
5α-Reductase 2 deficiency (5αR2D) is an autosomal recessive condition caused by a mutation in SRD5A2, a gene encoding the enzyme 5α-reductase type 2 (5αR2). The condition is rare, affects only genetic males, and has a broad spectrum.

Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive disorders characterized by impaired cortisol synthesis. It results from the deficiency of one of the five enzymes required for the synthesis of cortisol in the adrenal cortex. Most of these disorders involve excessive or deficient production of hormones such as glucocorticoids, mineralocorticoids, or sex steroids, and can alter development of primary or secondary sex characteristics in some affected infants, children, or adults. It is one of the most common autosomal recessive disorders in humans.

Lipoid congenital adrenal hyperplasia is an endocrine disorder that is an uncommon and potentially lethal form of congenital adrenal hyperplasia (CAH). It arises from defects in the earliest stages of steroid hormone synthesis: the transport of cholesterol into the mitochondria and the conversion of cholesterol to pregnenolone—the first step in the synthesis of all steroid hormones. Lipoid CAH causes mineralocorticoid deficiency in affected infants and children. Male infants are severely undervirilized causing their external genitalia to look feminine. The adrenals are large and filled with lipid globules derived from cholesterol.

Congenital adrenal hyperplasia due to 3β-hydroxysteroid dehydrogenase deficiency is an uncommon form of congenital adrenal hyperplasia (CAH) resulting from a mutation in the gene for one of the key enzymes in cortisol synthesis by the adrenal gland, 3β-hydroxysteroid dehydrogenase (3β-HSD) type II (HSD3B2). As a result, higher levels of 17α-hydroxypregnenolone appear in the blood with adrenocorticotropic hormone (ACTH) challenge, which stimulates adrenal corticosteroid synthesis.
Congenital adrenal hyperplasia due to 17α-hydroxylase deficiency is an uncommon form of congenital adrenal hyperplasia (CAH) resulting from a mutation in the gene CYP17A1, which produces the enzyme 17α-hydroxylase. It causes decreased synthesis of cortisol and sex hormones, with resulting increase in mineralocorticoid production. Thus, common symptoms include mild cortisol deficiency, ambiguous genitalia in men or amenorrhea at puberty in women, and hypokalemic hypertension. However, partial (incomplete) deficiency often has inconsistent symptoms between patients, and affected women may be asymptomatic except for infertility.
Kallmann syndrome (KS) is a genetic disorder that prevents a person from starting or fully completing puberty. Kallmann syndrome is a form of a group of conditions termed hypogonadotropic hypogonadism. To distinguish it from other forms of hypogonadotropic hypogonadism, Kallmann syndrome has the additional symptom of a total lack of sense of smell (anosmia) or a reduced sense of smell. If left untreated, people will have poorly defined secondary sexual characteristics, show signs of hypogonadism, almost invariably are infertile and are at increased risk of developing osteoporosis. A range of other physical symptoms affecting the face, hands and skeletal system can also occur.

Cytochrome P450 17A1 is an enzyme of the hydroxylase type that in humans is encoded by the CYP17A1 gene on chromosome 10. It is ubiquitously expressed in many tissues and cell types, including the zona reticularis and zona fasciculata of the adrenal cortex as well as gonadal tissues. It has both 17α-hydroxylase and 17,20-lyase activities, and is a key enzyme in the steroidogenic pathway that produces progestins, mineralocorticoids, glucocorticoids, androgens, and estrogens. More specifically, the enzyme acts upon pregnenolone and progesterone to add a hydroxyl (-OH) group at carbon 17 position (C17) of the steroid D ring, or acts upon 17α-hydroxyprogesterone and 17α-hydroxypregnenolone to split the side-chain off the steroid nucleus.

Estrogen insensitivity syndrome (EIS), or estrogen resistance, is a form of congenital estrogen deficiency or hypoestrogenism which is caused by a defective estrogen receptor (ER) – specifically, the estrogen receptor alpha (ERα) – that results in an inability of estrogen to mediate its biological effects in the body. Congenital estrogen deficiency can alternatively be caused by a defect in aromatase, the enzyme responsible for the biosynthesis of estrogens, a condition which is referred to as aromatase deficiency and is similar in symptomatology to EIS.

Disorders of sex development (DSDs), also known as differences in sex development or variations in sex characteristics (VSC), are congenital conditions affecting the reproductive system, in which development of chromosomal, gonadal, or anatomical sex is atypical. DSDs is a clinical term used in some medical settings for what are otherwise referred to as intersex traits. The term was first introduced in 2006 and has not been without controversy.

Aromatase deficiency is a rare condition characterized by extremely low levels or complete absence of the enzyme aromatase activity in the body. It is an autosomal recessive disease resulting from various mutations of gene CYP19 (P450arom) which can lead to ambiguous genitalia and delayed puberty in females, continued linear growth into adulthood and osteoporosis in males and virilization in pregnant mothers. As of 2020, fewer than 15 cases have been identified in genetically male individuals and at least 30 cases in genetically female individuals.
Hypergonadotropic hypogonadism (HH), also known as primary or peripheral/gonadal hypogonadism or primary gonadal failure, is a condition which is characterized by hypogonadism which is due to an impaired response of the gonads to the gonadotropins, follicle-stimulating hormone (FSH) and luteinizing hormone (LH), and in turn a lack of sex steroid production. As compensation and the lack of negative feedback, gonadotropin levels are elevated. Individuals with HH have an intact and functioning hypothalamus and pituitary glands so they are still able to produce FSH and LH. HH may present as either congenital or acquired, but the majority of cases are of the former nature. HH can be treated with hormone replacement therapy.
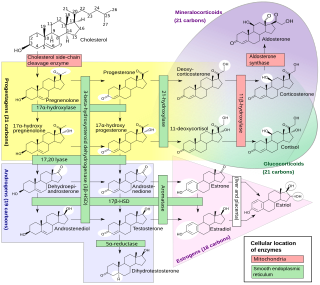
An inborn error of steroid metabolism is an inborn error of metabolism due to defects in steroid metabolism.
Leydig cell hypoplasia (LCH), also known as Leydig cell agenesis, is a rare autosomal recessive genetic and endocrine syndrome affecting an estimated 1 in 1,000,000 individuals with XY chromosomes. It is characterized by an inability of the body to respond to luteinizing hormone (LH), a gonadotropin which is normally responsible for signaling Leydig cells of the testicles to produce testosterone and other androgen sex hormones. The condition manifests itself as pseudohermaphroditism, hypergonadotropic hypogonadism, reduced or absent puberty, and infertility.
Gonadotropin-releasing hormone (GnRH) insensitivity also known as Isolated gonadotropin-releasing hormone (GnRH)deficiency (IGD) is a rare autosomal recessive genetic and endocrine syndrome which is characterized by inactivating mutations of the gonadotropin-releasing hormone receptor (GnRHR) and thus an insensitivity of the receptor to gonadotropin-releasing hormone (GnRH), resulting in a partial or complete loss of the ability of the gonads to synthesize the sex hormones. The condition manifests itself as isolated hypogonadotropic hypogonadism (IHH), presenting with symptoms such as delayed, reduced, or absent puberty, low or complete lack of libido, and infertility, and is the predominant cause of IHH when it does not present alongside anosmia.
Follicle-stimulating hormone (FSH) insensitivity, or ovarian insensitivity to FSH in females, also referable to as ovarian follicle hypoplasia or granulosa cell hypoplasia in females, is a rare autosomal recessive genetic and endocrine syndrome affecting both females and males, with the former presenting with much greater severity of symptomatology. It is characterized by a resistance or complete insensitivity to the effects of follicle-stimulating hormone (FSH), a gonadotropin which is normally responsible for the stimulation of estrogen production by the ovaries in females and maintenance of fertility in both sexes. The condition manifests itself as hypergonadotropic hypogonadism, reduced or absent puberty, amenorrhea, and infertility in females, whereas males present merely with varying degrees of infertility and associated symptoms.

Seviteronel is an experimental cancer medication which is under development by Viamet Pharmaceuticals and Innocrin Pharmaceuticals for the treatment of prostate cancer and breast cancer. It is a nonsteroidal CYP17A1 inhibitor and works by inhibiting the production of androgens and estrogens in the body. As of July 2017, seviteronel is in phase II clinical trials for both prostate cancer and breast cancer. In January 2016, it was designated fast-track status by the United States Food and Drug Administration for prostate cancer. In April 2017, seviteronel received fast-track designation for breast cancer as well.

A CYP17A1 inhibitor is a type of drug that inhibits the enzyme CYP17A1. CYP17A1 inhibitors work by blocking specific enzyme functions, impacting androgen biosynthesis.
An androgen synthesis inhibitor is a type of drug which inhibits the enzymatic synthesis of androgens, such as testosterone and dihydrotestosterone (DHT). They include:
Cytochrome b5 deficiency is a rare condition and form of isolated 17,20-lyase deficiency caused by deficiency in cytochrome b5, a small hemoprotein that acts as an allosteric factor to facilitate the interaction of CYP17A1 (17α-hydroxylase/17,20-lyase) with P450 oxidoreductase (POR), thereby allowing for the 17,20-lyase activity of CYP17A1. The condition affects both adrenal and gonadal androgen biosynthesis and results in male pseudohermaphroditism. The principal biological role of cytochrome b5 is reduction of methemoglobin, so cytochrome b5 deficiency can also result in elevated methemoglobin levels and/or methemoglobinemia, similarly to deficiency of cytochrome b5 reductase.

The androgen backdoor pathway is responsible for the synthesis of physiologically relevant androgens. This process starts with 21-carbon steroids, also known as pregnanes, and involves a step called "5α-reduction". Notably, this pathway does not require the intermediate formation of testosterone, hence the term "bypassing testosterone" is sometimes used in medical literature as the hallmark feature of this way of androgen biosynthesis. This feature is a key distinction from the conventional, canonical androgenic pathway, which necessitates the involvement of testosterone as an intermediate in the synthesis of androgens.
