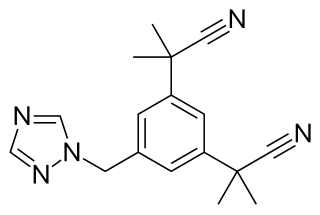
Anastrozole, sold under the brand name Arimidex among others, is an antiestrogenic medication used in addition to other treatments for breast cancer. Specifically it is used for hormone receptor-positive breast cancer. It has also been used to prevent breast cancer in those at high risk. It is taken by mouth.
Gonadotropin-releasing hormone (GnRH) is a releasing hormone responsible for the release of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) from the anterior pituitary. GnRH is a tropic peptide hormone synthesized and released from GnRH neurons within the hypothalamus. The peptide belongs to gonadotropin-releasing hormone family. It constitutes the initial step in the hypothalamic–pituitary–gonadal axis.
In medicine, precocious puberty is puberty occurring at an unusually early age. In most cases, the process is normal in every aspect except the unusually early age and simply represents a variation of normal development. There is early development of secondary sex characters and gametogenesis also starts earlier. Precocious puberty is of two types: true precocious puberty and pseudoprecocious puberty. In a minority of children with precocious puberty, the early development is triggered by a disease such as a tumor or injury of the brain.
Delayed puberty is when a person lacks or has incomplete development of specific sexual characteristics past the usual age of onset of puberty. The person may have no physical or hormonal signs that puberty has begun. In the United States, girls are considered to have delayed puberty if they lack breast development by age 13 or have not started menstruating by age 15. Boys are considered to have delayed puberty if they lack enlargement of the testicles by age 14. Delayed puberty affects about 2% of adolescents.
Gonadotropins are glycoprotein hormones secreted by gonadotropic cells of the anterior pituitary of vertebrates. This family includes the mammalian hormones follicle-stimulating hormone (FSH) and luteinizing hormone (LH), the placental/chorionic gonadotropins, human chorionic gonadotropin (hCG) and equine chorionic gonadotropin (eCG), as well as at least two forms of fish gonadotropins. These hormones are central to the complex endocrine system that regulates normal growth, sexual development, and reproductive function. LH and FSH are secreted by the anterior pituitary gland, while hCG and eCG are secreted by the placenta in pregnant women and mares, respectively. The gonadotropins act on the gonads, controlling gamete and sex hormone production.
Gonadarche refers to the earliest gonadal changes of puberty. In response to pituitary gonadotropins, the ovaries in females and the testes in males begin to grow and increase the production of the sex steroids, especially estradiol and testosterone. The ovary and testis have receptors, follicle cells and leydig cells, respectively, where gonadotropins bind to stimulate the maturation of the gonads and secretion of estrogen and testosterone. Certain disorders can result in changes to timing or nature of these processes.

Goserelin, sold under the brand name Zoladex among others, is a medication which is used to suppress production of the sex hormones, particularly in the treatment of breast cancer and prostate cancer. It is an injectable gonadotropin releasing hormone agonist.

Bicalutamide, sold under the brand name Casodex among others, is an antiandrogen medication that is primarily used to treat prostate cancer. It is typically used together with a gonadotropin-releasing hormone (GnRH) analogue or surgical removal of the testicles to treat metastatic prostate cancer (mPC). To a lesser extent, it is used at high doses for locally advanced prostate cancer (LAPC) as a monotherapy without castration. Bicalutamide was also previously used as monotherapy to treat localized prostate cancer (LPC), but authorization for this use was withdrawn following unfavorable trial findings. Besides prostate cancer, bicalutamide is limitedly used in the treatment of excessive hair growth and scalp hair loss in women, as a puberty blocker and component of feminizing hormone therapy for transgender girls and women, to treat gonadotropin-independent early puberty in boys, and to prevent overly long-lasting erections in men. It is taken by mouth.
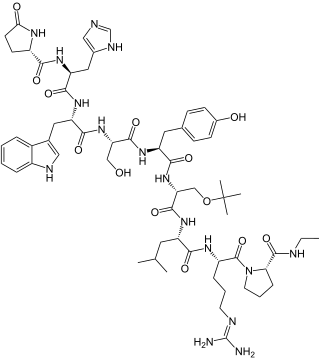
Buserelin, sold under the brand name Suprefact among others, is a medication which is used primarily in the treatment of prostate cancer and endometriosis. It is also used for other indications such as the treatment of premenopausal breast cancer, uterine fibroids, and early puberty, in assisted reproduction for female infertility, and as a part of transgender hormone therapy. In addition, buserelin is used in veterinary medicine. The medication is typically used as a nasal spray three times per day, but is also available for use as a solution or implant for injection into fat.

The hypothalamic–pituitary–gonadal axis refers to the hypothalamus, pituitary gland, and gonadal glands as if these individual endocrine glands were a single entity. Because these glands often act in concert, physiologists and endocrinologists find it convenient and descriptive to speak of them as a single system.

Triptorelin, sold under the brand name Decapeptyl among others, is a medication that acts as an agonist analog of gonadotropin-releasing hormone, repressing expression of luteinizing hormone (LH) and follicle-stimulating hormone (FSH).

A gonadotropin-releasing hormone agonist is a type of medication which affects gonadotropins and sex hormones. They are used for a variety of indications including in fertility medicine and to lower sex hormone levels in the treatment of hormone-sensitive cancers such as prostate cancer and breast cancer, certain gynecological disorders like heavy periods and endometriosis, high testosterone levels in women, early puberty in children, as a part of transgender hormone therapy, and to delay puberty in transgender youth among other uses. It is also used in the suppression of spontaneous ovulation as part of controlled ovarian hyperstimulation, an essential component in IVF. GnRH agonists are given by injections into fat, as implants placed into fat, and as nasal sprays.
The gonadotropin-releasing hormone receptor (GnRHR), also known as the luteinizing hormone releasing hormone receptor (LHRHR), is a member of the seven-transmembrane, G-protein coupled receptor (GPCR) family. It is the receptor of gonadotropin-releasing hormone (GnRH). Agonist binding to the GnRH receptor activates the Gq/11 family of heterotrimeric G proteins. The GnRHR is expressed on the surface of pituitary gonadotrope cells as well as lymphocytes, breast, ovary, and prostate.
The luteinizing hormone/choriogonadotropin receptor (LHCGR), also lutropin/choriogonadotropin receptor (LCGR) or luteinizing hormone receptor (LHR), is a transmembrane receptor found predominantly in the ovary and testis, but also many extragonadal organs such as the uterus and breasts. The receptor interacts with both luteinizing hormone (LH) and chorionic gonadotropins and represents a G protein-coupled receptor (GPCR). Its activation is necessary for the hormonal functioning during reproduction.
Hypergonadotropic hypogonadism (HH), also known as primary or peripheral/gonadal hypogonadism or primary gonadal failure, is a condition which is characterized by hypogonadism which is due to an impaired response of the gonads to the gonadotropins, follicle-stimulating hormone (FSH) and luteinizing hormone (LH), and in turn a lack of sex steroid production. As compensation and the lack of negative feedback, gonadotropin levels are elevated. Individuals with HH have an intact and functioning hypothalamus and pituitary glands so they are still able to produce FSH and LH. HH may present as either congenital or acquired, but the majority of cases are of the former nature. HH can be treated with hormone replacement therapy.

Leydig cell hypoplasia (LCH), also known as Leydig cell agenesis, is a rare autosomal recessive genetic and endocrine syndrome affecting an estimated 1 in 1,000,000 individuals with XY chromosomes. It is characterized by an inability of the body to respond to luteinizing hormone (LH), a gonadotropin which is normally responsible for signaling Leydig cells of the testicles to produce testosterone and other androgen sex hormones. The condition manifests itself as pseudohermaphroditism, hypergonadotropic hypogonadism, reduced or absent puberty, and infertility.
Gonadotropin-releasing hormone (GnRH) insensitivity also known as Isolated gonadotropin-releasing hormone (GnRH)deficiency (IGD) is a rare autosomal recessive genetic and endocrine syndrome which is characterized by inactivating mutations of the gonadotropin-releasing hormone receptor (GnRHR) and thus an insensitivity of the receptor to gonadotropin-releasing hormone (GnRH), resulting in a partial or complete loss of the ability of the gonads to synthesize the sex hormones. The condition manifests itself as isolated hypogonadotropic hypogonadism (IHH), presenting with symptoms such as delayed, reduced, or absent puberty, low or complete lack of libido, and infertility, and is the predominant cause of IHH when it does not present alongside anosmia.

A progonadotropin, or hypergonadotropin, also known as a gonad stimulant, is a type of drug which increases the secretion of one or both of the major gonadotropins, luteinizing hormone (LH) and follicle-stimulating hormone (FSH). This, in turn, results in increased function and maintenance of the gonads and increased gonadal steroidogenesis of sex hormones such as androgens, estrogens, and progestogens. Progonadotropins are the functional opposites of antigonadotropins. They have clinical applications in the treatment of hypogonadism and infertility. Conversely, hypergonadotropic effects can occur as a side effect of some drugs. Examples of progonadotropic drugs include gonadotropin-releasing hormone (GnRH) agonists when administered in a pulsatile manner, antiestrogens such as tamoxifen, clomifene, fulvestrant, and aromatase inhibitors like anastrozole, and, only in men, pure antiandrogens such as flutamide, bicalutamide, enzalutamide, and apalutamide.
The medical uses of bicalutamide, a nonsteroidal antiandrogen (NSAA), include the treatment of androgen-dependent conditions and hormone therapy to block the effects of androgens. Indications for bicalutamide include the treatment of prostate cancer in men, skin and hair conditions such as acne, seborrhea, hirsutism, and pattern hair loss in women, high testosterone levels in women, hormone therapy in transgender women, as a puberty blocker to prevent puberty in transgender girls and to treat early puberty in boys, and the treatment of long-lasting erections in men. It may also have some value in the treatment of paraphilias and hypersexuality in men.
The pharmacology of bicalutamide is the study of the pharmacodynamic and pharmacokinetic properties of the nonsteroidal antiandrogen (NSAA) bicalutamide. In terms of pharmacodynamics, bicalutamide acts as a selective antagonist of the androgen receptor (AR), the biological target of androgens like testosterone and dihydrotestosterone (DHT). It has no capacity to activate the AR. It does not decrease androgen levels and has no other important hormonal activity. The medication has progonadotropic effects due to its AR antagonist activity and can increase androgen, estrogen, and neurosteroid production and levels. This results in a variety of differences of bicalutamide monotherapy compared to surgical and medical castration, such as indirect estrogenic effects and associated benefits like preservation of sexual function and drawbacks like gynecomastia. Bicalutamide can paradoxically stimulate late-stage prostate cancer due to accumulated mutations in the cancer. When used as a monotherapy, bicalutamide can induce breast development in males due to its estrogenic effects. Unlike other kinds of antiandrogens, it may have less adverse effect on the testes and fertility.
