Leydig cell hypoplasia does not occur in people with XX chromosomes as they do not have either Leydig cells or testicles. However, the cause of the condition in people with XY chromosomes, luteinizing hormone insensitivity, does affect people with XX chromosomes, and because LH plays a role in the female reproductive system, it can result in primary amenorrhea or oligomenorrhea (absent or reduced menstruation), infertility due to anovulation, and ovarian cysts.[2][4]
A related condition is follicle-stimulating hormone (FSH) insensitivity, which presents with similar symptoms to those of Leydig cell hypoplasia but with the symptoms in the respective sexes reversed (i.e., hypogonadism and sexual infantilism in XX individuals and difficulties with fertility in XY individuals). Despite their similar causes, FSH insensitivity is considerably less common in comparison to LH insensitivity.[5]
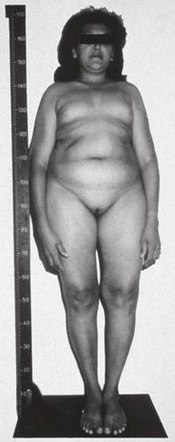
A 46,XY (genetically "male") woman with complete insensitivity to LH, resulting in fully feminized genitalia at birth. Puberty appears to have commenced, likely due to estrogen replacement therapy.
Leydig cell hypoplasia is caused by genetic mutations in LHCGR, a gene which encodes the LH/hCG receptor. LH normally acts through the LH/hCG receptor to stimulate the growth of Leydig cells in the testicles and the production of androgens such as testosterone and dihydrotestosterone (DHT) by these cells. In Leydig cell hypoplasia however, there is a reduced capacity for the LH/hCG receptor to respond to LH. This results in hypoplasia or absence of Leydig cells, testicular atrophy, and lower than normal androgen levels. In the most severe form of the condition in which there is a complete lack of response of the Leydig cells to LH, androgen production by the testicles is virtually negligible and secondary sexual characteristics entirely fail to develop at puberty.[2][3][7][8][9]
People with Leydig cell hypoplasia type I display no response to the hCG stimulation test; there is no increase in serum levels of testosterone and dihydrotestosterone.[10] Leydig cell hypoplasia type II can display either a pronounced rise of testosterone levels or no rise. [citation needed]
In any case, the diagnosis is confirmed on biopsy of the testes, revealing either absent or hypoplastic Leydig cells. The inside of the testis will be grayish and mucous, displaying arrested spermatogenesis and the presence of Sertoli cells.[11] The diagnosis can also be confirmed by looking for mutations in the gene for the LH receptor.[12]
A diagnosis of Leydig cell hypoplasia is usually made in the neonatal period, following the discovery of ambiguous genitalia, or at puberty, when secondary sex characteristics fail to develop. Puberty is the most common time for Leydig cell hypoplasia to be diagnosed.[11][13]
Treatment
Patients with Leydig cell hypoplasia may be treated with hormone replacement therapy (i.e., with androgens), which will result in normal sexual development and the resolution of most symptoms. In the case of 46,XY (genetically "male") individuals who are phenotypically female and/or have a female gender identity, estrogens are recommended instead. Surgical correction of the genitals in individuals with male gender identity may be required, and, if necessary, an orchidopexy (relocation of the undescended testes to the scrotum) may be performed as well.[3]
↑ Mendonca BB, Costa EM, Belgorosky A, Rivarola MA, Domenice S (April 2010). "46,XY DSD due to impaired androgen production". Best Practice & Research. Clinical Endocrinology & Metabolism. 24 (2): 243–62. doi:10.1016/j.beem.2009.11.003. hdl:11336/98827. PMID20541150.
↑ Latronico AC, Arnhold IJ (September 2006). "Inactivating mutations of LH and FSH receptors--from genotype to phenotype". Pediatric Endocrinology Reviews. 4 (1): 28–31. PMID17021580.
↑ Amesse, Lawrence S. & Pfaff-Amesse, Teresa (2007). "Chapter 12: Congenital Anomalies of the Female Reproductive Tract". In Falcone, Tomasso & Hurd, William W. (eds.). Clinical Reproductive Medicine and Surgery. Elsevier Health Sciences. p.184. ISBN978-0-323-03309-1.
1 2 Nistal, Manuel & González-Peramato, Pilar (2016). "Congenital Lesions". In Colecchia, Maurizio (ed.). Pathology of Testicular and Penile Neoplasms. Springer. p.184. ISBN978-3-319-27617-5.
↑ Nieschlag, Eberhard & Behre, Hermann (June 29, 2013), "Chapter 8: Disorders at the Testicular Level", Andrology: Male Reproductive Health and Dysfunction, Springer Science & Business Media, p.166, ISBN978-3-662-04491-9
↑ McCann-Crosby, Bonnie & Sutton, V. Reid (2015). "Disorders of Sexual Development". In Gambello, Michael J. & Sutton, V. Reid (eds.). Genetics Diagnosis, Inborn Errors of Metabolism and Newborn Screening: An Update. Elsevier Health Sciences. p.407. ISBN978-0-323-35685-5.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.