In cellular biology, paracrine signaling is a form of cell signaling, a type of cellular communication in which a cell produces a signal to induce changes in nearby cells, altering the behaviour of those cells. Signaling molecules known as paracrine factors diffuse over a relatively short distance, as opposed to cell signaling by endocrine factors, hormones which travel considerably longer distances via the circulatory system; juxtacrine interactions; and autocrine signaling. Cells that produce paracrine factors secrete them into the immediate extracellular environment. Factors then travel to nearby cells in which the gradient of factor received determines the outcome. However, the exact distance that paracrine factors can travel is not certain.

Crouzon syndrome is an autosomal dominant genetic disorder known as a branchial arch syndrome. Specifically, this syndrome affects the first branchial arch, which is the precursor of the maxilla and mandible. Since the branchial arches are important developmental features in a growing embryo, disturbances in their development create lasting and widespread effects.

Apert syndrome is a form of acrocephalosyndactyly, a congenital disorder characterized by malformations of the skull, face, hands and feet. It is classified as a branchial arch syndrome, affecting the first branchial arch, the precursor of the maxilla and mandible. Disturbances in the development of the branchial arches in fetal development create lasting and widespread effects.

Jackson–Weiss syndrome (JWS) is a genetic disorder characterized by foot abnormalities and the premature fusion of certain bones of the skull (craniosynostosis), which prevents further growth of the skull and affects the shape of the head and face. This genetic disorder can also sometimes cause intellectual disability and crossed eyes. It was characterized in 1976.

Pfeiffer syndrome is a rare genetic disorder, characterized by the premature fusion of certain bones of the skull (craniosynostosis), which affects the shape of the head and face. The syndrome includes abnormalities of the hands and feet, such as wide and deviated thumbs and big toes.
Fibroblast growth factors (FGF) are a family of cell signalling proteins produced by macrophages; they are involved in a wide variety of processes, most notably as crucial elements for normal development in animal cells. Any irregularities in their function lead to a range of developmental defects. These growth factors typically act as systemic or locally circulating molecules of extracellular origin that activate cell surface receptors. A defining property of FGFs is that they bind to heparin and to heparan sulfate. Thus, some are sequestered in the extracellular matrix of tissues that contains heparan sulfate proteoglycans and are released locally upon injury or tissue remodeling.

Fibroblast growth factor 1, (FGF-1) also known as acidic fibroblast growth factor (aFGF), is a growth factor and signaling protein encoded by the FGF1 gene. It is synthesized as a 155 amino acid polypeptide, whose mature form is a non-glycosylated 17-18 kDa protein. Fibroblast growth factor protein was first purified in 1975, but soon afterwards others using different conditions isolated acidic FGF, Heparin-binding growth factor-1, and Endothelial cell growth factor-1. Gene sequencing revealed that this group was actually the same growth factor and that FGF1 was a member of a family of FGF proteins.

Receptor tyrosine kinases (RTKs) are the high-affinity cell surface receptors for many polypeptide growth factors, cytokines, and hormones. Of the 90 unique tyrosine kinase genes identified in the human genome, 58 encode receptor tyrosine kinase proteins. Receptor tyrosine kinases have been shown not only to be key regulators of normal cellular processes but also to have a critical role in the development and progression of many types of cancer. Mutations in receptor tyrosine kinases lead to activation of a series of signalling cascades which have numerous effects on protein expression. Receptor tyrosine kinases are part of the larger family of protein tyrosine kinases, encompassing the receptor tyrosine kinase proteins which contain a transmembrane domain, as well as the non-receptor tyrosine kinases which do not possess transmembrane domains.

SMAD4, also called SMAD family member 4, Mothers against decapentaplegic homolog 4, or DPC4 is a highly conserved protein present in all metazoans. It belongs to the SMAD family of transcription factor proteins, which act as mediators of TGF-β signal transduction. The TGFβ family of cytokines regulates critical processes during the lifecycle of metazoans, with important roles during embryo development, tissue homeostasis, regeneration, and immune regulation.

Activin A receptor, type I (ACVR1) is a protein which in humans is encoded by the ACVR1 gene; also known as ALK-2. ACVR1 has been linked to the 2q23-24 region of the genome. This protein is important in the bone morphogenic protein (BMP) pathway which is responsible for the development and repair of the skeletal system. While knock-out models with this gene are in progress, the ACVR1 gene has been connected to fibrodysplasia ossificans progressiva, a very rare progressive genetic disease characterized by heterotopic ossification of muscles, tendons and ligaments. It is a bone morphogenetic protein receptor, type 1.
The fibroblast growth factor receptors (FGFR) are, as their name implies, receptors that bind to members of the fibroblast growth factor (FGF) family of proteins. Some of these receptors are involved in pathological conditions. For example, a point mutation in FGFR3 can lead to achondroplasia.
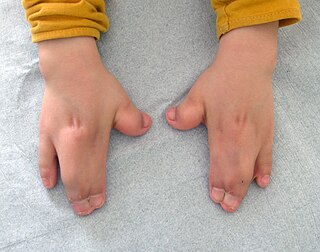
Acrocephalosyndactyly is a group of congenital conditions characterized by irregular features of the face and skull (craniosynostosis) and hands and feet (syndactyly). Craniosynostosis occurs when the cranial sutures, the fibrous tissue connecting the skull bones, fuse the cranial bones early in development. Cranial sutures allow the skull bones to continue growing until they fuse at age 24. Premature fusing of the cranial sutures can result in alterations to the skull shape and interfere with brain growth. Syndactyly occurs when digits of the hands or feet are fused together. When polydactyly is also present, the classification is acrocephalopolysyndactyly. Polydactyly occurs when the hands or feet possess additional digits. Acrocephalosyndactyly is usually diagnosed after birth, although prenatal diagnosis is sometimes possible if the genetic variation is present in family members, as the conditions are typically inherited in an autosomal dominant pattern Treatment often involves surgery in early childhood to correct for craniosynostosis and syndactyly.

Fibroblast growth factor receptor 1 (FGFR1), also known as basic fibroblast growth factor receptor 1, fms-related tyrosine kinase-2 / Pfeiffer syndrome, and CD331, is a receptor tyrosine kinase whose ligands are specific members of the fibroblast growth factor family. FGFR1 has been shown to be associated with Pfeiffer syndrome, and clonal eosinophilias.

Fibroblast growth factor receptor 3 is a protein that in humans is encoded by the FGFR3 gene. FGFR3 has also been designated as CD333. The gene, which is located on chromosome 4, location p16.3, is expressed in tissues such as the cartilage, brain, intestine, and kidneys.

Fibroblast growth factor receptor 4 is a protein that in humans is encoded by the FGFR4 gene. FGFR4 has also been designated as CD334.

Fibroblast growth factor 10 is a protein that in humans is encoded by the FGF10 gene.

Fibroblast growth factor 8(FGF-8) is a protein that in humans is encoded by the FGF8 gene.

Fibroblast growth factor receptor-like 1 is a protein that in humans is encoded by the FGFRL1 gene.
Beare–Stevenson cutis gyrata syndrome is a rare genetic disorder characterized by craniosynostosis and a specific skin abnormality, called cutis gyrata, characterized by a furrowed and wrinkled appearance ; thick, dark, velvety areas of skin are sometimes found on the hands and feet and in the groin.
Fibroblast growth factor receptor oncogene partner 2 (FGFR1OP2) was identified in a study on myeloproliferative syndrome (EMS). The study aimed to identify the partner genes to the fibroblast growth factor receptor 1 (FGFR1) involved in the syndrome. Using the 5'-RACE PCR technique, FGFR1OP2 was identified as a novel gene with no known function.