
A protein kinase is a kinase which selectively modifies other proteins by covalently adding phosphates to them (phosphorylation) as opposed to kinases which modify lipids, carbohydrates, or other molecules. Phosphorylation usually results in a functional change of the target protein (substrate) by changing enzyme activity, cellular location, or association with other proteins. The human genome contains about 500 protein kinase genes and they constitute about 2% of all human genes. There are two main types of protein kinase. The great majority are serine/threonine kinases, which phosphorylate the hydroxyl groups of serines and threonines in their targets. Most of the others are tyrosine kinases, although additional types exist. Protein kinases are also found in bacteria and plants. Up to 30% of all human proteins may be modified by kinase activity, and kinases are known to regulate the majority of cellular pathways, especially those involved in signal transduction.
A tyrosine kinase is an enzyme that can transfer a phosphate group from ATP to the tyrosine residues of specific proteins inside a cell. It functions as an "on" or "off" switch in many cellular functions.

Gefitinib, sold under the brand name Iressa, is a medication used for certain breast, lung and other cancers. Gefitinib is an EGFR inhibitor, like erlotinib, which interrupts signaling through the epidermal growth factor receptor (EGFR) in target cells. Therefore, it is only effective in cancers with mutated and overactive EGFR, but resistances to gefitinib can arise through other mutations. It is marketed by AstraZeneca and Teva.

The restriction point (R), also known as the Start or G1/S checkpoint, is a cell cycle checkpoint in the G1 phase of the animal cell cycle at which the cell becomes "committed" to the cell cycle, and after which extracellular signals are no longer required to stimulate proliferation. The defining biochemical feature of the restriction point is the activation of G1/S- and S-phase cyclin-CDK complexes, which in turn phosphorylate proteins that initiate DNA replication, centrosome duplication, and other early cell cycle events. It is one of three main cell cycle checkpoints, the other two being the G2-M DNA damage checkpoint and the spindle checkpoint.

The epidermal growth factor receptor is a transmembrane protein that is a receptor for members of the epidermal growth factor family of extracellular protein ligands.

Hepatocyte growth factor receptor is a protein that in humans is encoded by the MET gene. The protein possesses tyrosine kinase activity. The primary single chain precursor protein is post-translationally cleaved to produce the alpha and beta subunits, which are disulfide linked to form the mature receptor.
Biological crosstalk refers to instances in which one or more components of one signal transduction pathway affects another. This can be achieved through a number of ways with the most common form being crosstalk between proteins of signaling cascades. In these signal transduction pathways, there are often shared components that can interact with either pathway. A more complex instance of crosstalk can be observed with transmembrane crosstalk between the extracellular matrix (ECM) and the cytoskeleton.
Mitogen Activated Protein (MAP) kinase kinase kinase is a serine/threonine-specific protein kinase which acts upon MAP kinase kinase. Subsequently, MAP kinase kinase activates MAP kinase. Several types of MAPKKK can exist but are mainly characterized by the MAP kinases they activate. MAPKKKs are stimulated by a large range of stimuli, primarily environmental and intracellular stressors. MAPKKK is responsible for various cell functions such as cell proliferation, cell differentiation, and apoptosis. The duration and intensity of signals determine which pathway ensues. Additionally, the use of protein scaffolds helps to place the MAPKKK in close proximity with its substrate to allow for a reaction. Lastly, because MAPKKK is involved in a series of several pathways, it has been used as a therapeutic target for cancer, amyloidosis, and neurodegenerative diseases. In humans, there are at least 19 genes which encode MAP kinase kinase kinases:
The MAPK/ERK pathway is a chain of proteins in the cell that communicates a signal from a receptor on the surface of the cell to the DNA in the nucleus of the cell.

Receptor tyrosine kinases (RTKs) are the high-affinity cell surface receptors for many polypeptide growth factors, cytokines, and hormones. Of the 90 unique tyrosine kinase genes identified in the human genome, 58 encode receptor tyrosine kinase proteins. Receptor tyrosine kinases have been shown not only to be key regulators of normal cellular processes but also to have a critical role in the development and progression of many types of cancer. Mutations in receptor tyrosine kinases lead to activation of a series of signalling cascades which have numerous effects on protein expression. The receptors are generally activated by dimerization and substrate presentation. Receptor tyrosine kinases are part of the larger family of protein tyrosine kinases, encompassing the receptor tyrosine kinase proteins which contain a transmembrane domain, as well as the non-receptor tyrosine kinases which do not possess transmembrane domains.

Platelet-derived growth factor receptors (PDGF-R) are cell surface tyrosine kinase receptors for members of the platelet-derived growth factor (PDGF) family. PDGF subunits -A and -B are important factors regulating cell proliferation, cellular differentiation, cell growth, development and many diseases including cancer. There are two forms of the PDGF-R, alpha and beta each encoded by a different gene. Depending on which growth factor is bound, PDGF-R homo- or heterodimerizes.
In molecular biology, extracellular signal-regulated kinases (ERKs) or classical MAP kinases are widely expressed protein kinase intracellular signalling molecules that are involved in functions including the regulation of meiosis, mitosis, and postmitotic functions in differentiated cells. Many different stimuli, including growth factors, cytokines, virus infection, ligands for heterotrimeric G protein-coupled receptors, transforming agents, and carcinogens, activate the ERK pathway.
Matuzumab is a humanized monoclonal antibody for the treatment of cancer. It binds to the epidermal growth factor receptor (EGFR) with high affinity. The mouse monoclonal antibody (mAb425) from which matuzumab was developed at the Wistar Institute in Philadelphia, Pennsylvania
Ras-GRF1 is a guanine nucleotide exchange factor. Its function is to release guanosine diphosphate, GDP, from the signaling protein RAS, thus increasing the activity of RAS by allowing it to bind to guanosine triphosphate, GTP, returning it to its active state. In this way, Ras-GRF1 has a key role in regulating the RAS signaling pathway. Ras-GRF1 mediates the activation of RAS via Ca2+ bound calmodulin protein.
A growth factor receptor is a receptor that binds to a growth factor. Growth factor receptors are the first stop in cells where the signaling cascade for cell differentiation and proliferation begins. Growth factors, which are ligands that bind to the receptor are the initial step to activating the growth factor receptors and tells the cell to grow and/or divide.
The ErbB family of proteins contains four receptor tyrosine kinases, structurally related to the epidermal growth factor receptor (EGFR), its first discovered member. In humans, the family includes Her1, Her2 (ErbB2), Her3 (ErbB3), and Her4 (ErbB4). The gene symbol, ErbB, is derived from the name of a viral oncogene to which these receptors are homologous: erythroblastic leukemia viral oncogene. Insufficient ErbB signaling in humans is associated with the development of neurodegenerative diseases, such as multiple sclerosis and Alzheimer's disease, while excessive ErbB signaling is associated with the development of a wide variety of types of solid tumor.

SHC-transforming protein 1 is a protein that in humans is encoded by the SHC1 gene. SHC has been found to be important in the regulation of apoptosis and drug resistance in mammalian cells.

Receptor tyrosine-protein kinase erbB-3, also known as HER3, is a membrane bound protein that in humans is encoded by the ERBB3 gene.
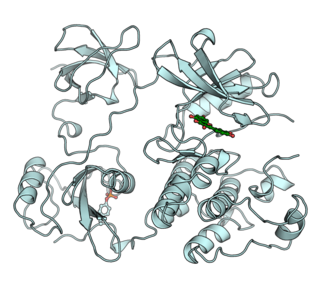
Activated CDC42 kinase 1, also known as ACK1, is an enzyme that in humans is encoded by the TNK2 gene. TNK2 gene encodes a non-receptor tyrosine kinase, ACK1, that binds to multiple receptor tyrosine kinases e.g. EGFR, MERTK, AXL, HER2 and insulin receptor (IR). ACK1 also interacts with Cdc42Hs in its GTP-bound form and inhibits both the intrinsic and GTPase-activating protein (GAP)-stimulated GTPase activity of Cdc42Hs. This binding is mediated by a unique sequence of 47 amino acids C-terminal to an SH3 domain. The protein may be involved in a regulatory mechanism that sustains the GTP-bound active form of Cdc42Hs and which is directly linked to a tyrosine phosphorylation signal transduction pathway. Several alternatively spliced transcript variants have been identified from this gene, but the full-length nature of only two transcript variants has been determined.
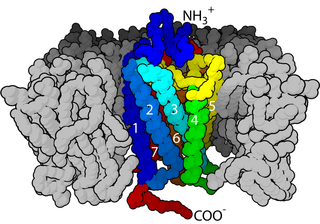
Cell surface receptors are receptors that are embedded in the plasma membrane of cells. They act in cell signaling by receiving extracellular molecules. They are specialized integral membrane proteins that allow communication between the cell and the extracellular space. The extracellular molecules may be hormones, neurotransmitters, cytokines, growth factors, cell adhesion molecules, or nutrients; they react with the receptor to induce changes in the metabolism and activity of a cell. In the process of signal transduction, ligand binding affects a cascading chemical change through the cell membrane.