Ohtahara syndrome (OS), also known as early infantile developmental and epileptic encephalopathy (EIDEE)[2] is a progressiveepilepticencephalopathy. The syndrome is outwardly characterized by tonic spasms and partial seizures within the first few months of life,[3] and receives its more elaborate name from the pattern of burst activity on an electroencephalogram (EEG). It is an extremely debilitating progressive neurological disorder, involving intractable seizures and severe intellectual disabilities. No single cause has been identified, although in many cases structural brain damage is present.[4]
Ohtahara syndrome is rare and the earliest-appearing age-related epileptic encephalopathy, with seizure onset occurring within the first three months of life, and often in the first ten days.[5] Many, but not all, cases of OS evolve into other seizure disorders, namely West syndrome and Lennox-Gastaut syndrome.[4]
The primary outward manifestation of OS is seizures, usually presenting as tonic seizures (a generalized seizure involving a sudden stiffening of the limbs).[6] Other seizure types that may occur include focal seizures, clusters of infantile spasms, and, rarely, myoclonic seizures. In addition to seizures, children with OS exhibit profound mental and physical disabilities.[citation needed]
Clinically, OS is characterized by a "burst suppression" pattern on an EEG. This pattern involves high voltage spike wave discharge followed by little brain wave activity.[4]
It is named for the Japanese neurologist Shunsuke Ohtahara (1930–2013), who identified it in 1976.[5]
Signs and symptoms
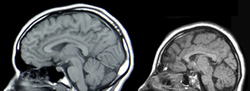
MRIs of a normal individual (left) and a patient with microcephaly caused by genetic mutation (right)
Both female and male infants born with OS may experience symptoms while asleep or awake. Many children die from OS within their first 2 years of life, while those who survive maintain physical and cognitive disabilities such as excessive fatigue, difficulty feeding, chest infections and slow developmental progress.[2][7] Although birth history and head size of infants is typically normal, microcephaly may occur.[3] Certain genetic variants manifest with additional signs such as dyskinetic movements and an atypical Rett-syndrome appearance.[8]
No single cause of OS has been identified. In most cases, there is severe atrophy of both hemispheres of the brain. Cerebral malformations such as hemimegalencephaly, porencephaly, Aicardi syndrome, olivary-dentate dysplasia, agenesis of mamillary bodies, linear sebaceous nevus syndrome, cerebral dysgenesis, and focal cortical dysplasia have been noted as suspect causes.[9]
Less often, the root of the disorder is an underlying metabolic syndrome, though mitochondrial disorders, non-ketotic hyperglycinemia, and enzyme deficiency remain elusive as causes. Their mechanisms are not entirely known.[3]
Diagnosis
Electroencephalogram (EEG) displaying burst suppression patterns. Onset of bursts are indicated by solid arrows; offset, by open arrows. In both A and B, the interval between each vertical dotted line is one second
The diagnosis is based on the clinical presentation and on typical electroencephalographic patterns based on time of onset.[24][2] Typically, onset of seizures and spasms have been indicative of OS diagnosis, while MRI and abnormal EEG "burst suppression" pattern can confirm. Genetic testing with chromosomal microarray analysis followed by an epilepsy gene panel or whole exome sequencing may be considered after MRI imaging has been exhausted.[25][26]
Treatment outlook is poor. Anticonvulsant drugs and glucocorticoid steroids may be used to try to control the seizures, but their effectiveness is limited. Most therapies are related to symptoms and day-to-day living.[4] For cases related to focal brain lesions, epilepsy surgery or functional hemispherectomy may be considered.[7][3][28] Risk factors include infection, blood loss, loss of vision, speech, memory, or movement.[citation needed]
Therapy for those with OS are based on severity of seizure activity and are supportive in nature. This may include treatment for abnormal muscle tone, stomach or lung problems.[7] A ketogenic diet may be suggested for reduction of symptoms.[2] Should the child survive past the age of three, vagus nerve stimulation could be considered.[2] No recent findings allude to preventive methods for pregnant mothers.[citation needed]
Prognosis
Prognosis is poor for infants with OS, and can be characterized by management of seizures, effects of secondary symptoms and shortened life span (up to 3 years of age). Survivors have severe psychomotor impairments and are dependent on their caretaker for support. Family members of infants with OS may consult with a palliative care team as symptoms may worsen or develop. Death is often due to strain from seizure activity, pneumonia or other complications from motor disabilities.[8]
Prospects of recovering from OS after hemispherectomy surgery has been shown to be favorable, with patients experiencing "catch up" in development.[29]
Epidemiology
Incidence has been estimated at 1/100 000 births in Japan and 1/50,000 births in the U.K.[30] Approximately 100 cases total have been reported but this may be an underestimate. since OS neonates with early death may escape clinico-EEG diagnosis.[9] Male cases slightly predominate those of females.
Ivan Cameron, son of David Cameron, former leader of the British Conservative Party and Prime Minister of the UK, was born with the condition and cerebral palsy. He died aged six on 25 February 2009, while his father was still opposition leader.[32]
Dr William H. Thomas, a United States doctor, has two daughters with this condition. He spoke about them during a PBS interview.[33]
1 2 Ohtahara S, Ishida T, Oka E, etal. (1976). "特異な年齢依存性てんかん性脳症the early-infantile epileptic encephalopathy with suppression-burstに関する研究" [On the specific age dependent epileptic syndrome: the early-infantile epileptic encephalopathy with suppression-burst]. No to Hattatsu (in Japanese). 8 (4): 270–80. doi:10.11251/ojjscn1969.8.270.
↑ Holmes, Gregory L. (January 2004). "Tonic". Epilepsy.com/Professionals. Retrieved 2007-11-26.
↑ Malik, Saleem I.; Galliani, Carlos A.; Hernandez, Angel W.; Donahue, David J. (December 2013). "Epilepsy surgery for early infantile epileptic encephalopathy (ohtahara syndrome)". Journal of Child Neurology. 28 (12): 1607–1617. doi:10.1177/0883073812464395. ISSN1708-8283. PMID23143728. S2CID21646215.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.