This article needs more reliable medical references for verification or relies too heavily on primary sources .(October 2019) |  |
Gonadotropin insensitivity includes:
This article needs more reliable medical references for verification or relies too heavily on primary sources .(October 2019) | |
Gonadotropin insensitivity includes:
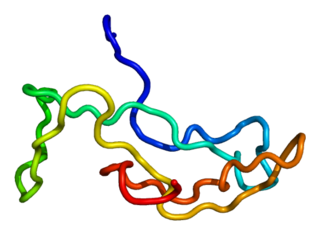
Luteinizing hormone is a hormone produced by gonadotropic cells in the anterior pituitary gland. The production of LH is regulated by gonadotropin-releasing hormone (GnRH) from the hypothalamus. In females, an acute rise of LH known as an LH surge, triggers ovulation and development of the corpus luteum. In males, where LH had also been called interstitial cell–stimulating hormone (ICSH), it stimulates Leydig cell production of testosterone. It acts synergistically with follicle-stimulating hormone (FSH).
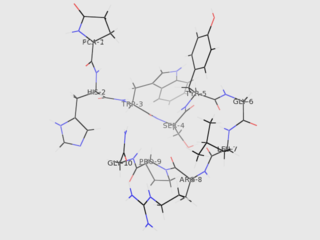
Gonadotropin-releasing hormone (GnRH) is a releasing hormone responsible for the release of follicle-stimulating hormone (FSH) and luteinizing hormone (LH) from the anterior pituitary. GnRH is a tropic peptide hormone synthesized and released from GnRH neurons within the hypothalamus. The peptide belongs to gonadotropin-releasing hormone family. It constitutes the initial step in the hypothalamic–pituitary–gonadal axis.
Gonadotropins are glycoprotein hormones secreted by gonadotropic cells of the anterior pituitary of vertebrates. This family includes the mammalian hormones follicle-stimulating hormone (FSH) and luteinizing hormone (LH), the placental/chorionic gonadotropins, human chorionic gonadotropin (hCG) and equine chorionic gonadotropin (eCG), as well as at least two forms of fish gonadotropins. These hormones are central to the complex endocrine system that regulates normal growth, sexual development, and reproductive function. LH and FSH are secreted by the anterior pituitary gland, while hCG and eCG are secreted by the placenta in pregnant humans and mares, respectively. The gonadotropins act on the gonads, controlling gamete and sex hormone production.
Hypogonadism means diminished functional activity of the gonads—the testicles or the ovaries—that may result in diminished production of sex hormones. Low androgen levels are referred to as hypoandrogenism and low estrogen as hypoestrogenism. These are responsible for the observed signs and symptoms in both males and females.
Isolated hypogonadotropic hypogonadism (IHH), also called idiopathic or congenital hypogonadotropic hypogonadism (CHH), as well as isolated or congenital gonadotropin-releasing hormone deficiency (IGD), is a condition which results in a small subset of cases of hypogonadotropic hypogonadism (HH) due to deficiency in or insensitivity to gonadotropin-releasing hormone (GnRH) where the function and anatomy of the anterior pituitary is otherwise normal and secondary causes of HH are not present.

Gonadotropin-releasing hormone antagonists are a class of medications that antagonize the gonadotropin-releasing hormone receptor and thus the action of gonadotropin-releasing hormone (GnRH). They are used in the treatment of prostate cancer, endometriosis, uterine fibroids, female infertility in assisted reproduction, and for other indications.

Familial male-limited precocious puberty, often abbreviated as FMPP, also known as familial sexual precocity or gonadotropin-independent testotoxicosis, is a form of gonadotropin-independent precocious puberty in which boys experience early onset and progression of puberty. Signs of puberty can develop as early as an age of 1 year.
The gonadotropin-releasing hormone receptor (GnRHR), also known as the luteinizing hormone releasing hormone receptor (LHRHR), is a member of the seven-transmembrane, G-protein coupled receptor (GPCR) family. It is the receptor of gonadotropin-releasing hormone (GnRH). The GnRHR is expressed on the surface of pituitary gonadotrope cells as well as lymphocytes, breast, ovary, and prostate.

Estrogen insensitivity syndrome (EIS), or estrogen resistance, is a form of congenital estrogen deficiency or hypoestrogenism which is caused by a defective estrogen receptor (ER) – specifically, the estrogen receptor alpha (ERα) – that results in an inability of estrogen to mediate its biological effects in the body. Congenital estrogen deficiency can alternatively be caused by a defect in aromatase, the enzyme responsible for the biosynthesis of estrogens, a condition which is referred to as aromatase deficiency and is similar in symptomatology to EIS.

Gonadotropin-releasing hormone receptor is a protein that in humans is encoded by the GNRHR gene.

Putative gonadotropin-releasing hormone II receptor is a protein that in humans is encoded by the GNRHR2 gene.

A micropenis is an unusually small penis. A common criterion is a dorsal penile length of at least 2.5 standard deviations smaller than the mean human penis size. A micropenis is stretched penile length equal to or less than 1.9 cm in term infants, and 9.3 cm in adults. The condition is usually recognized shortly after birth. The term is most often used medically when the rest of the penis, scrotum, and perineum are without ambiguity, such as hypospadias. A microphallus describes a medical term where other sections of genitallia are different, such as hypospadias or cryptorchidism. Micropenis incidence is about 1.5 in 10,000 male newborns in North America.
Hypergonadotropic hypogonadism (HH), also known as primary or peripheral/gonadal hypogonadism or primary gonadal failure, is a condition which is characterized by hypogonadism which is due to an impaired response of the gonads to the gonadotropins, follicle-stimulating hormone (FSH) and luteinizing hormone (LH), and in turn a lack of sex steroid production. As compensation and the lack of negative feedback, gonadotropin levels are elevated. Individuals with HH have an intact and functioning hypothalamus and pituitary glands so they are still able to produce FSH and LH. HH may present as either congenital or acquired, but the majority of cases are of the former nature. HH can be treated with hormone replacement therapy.
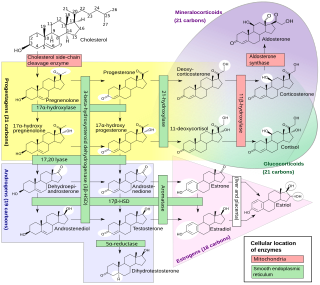
An inborn error of steroid metabolism is an inborn error of metabolism due to defects in steroid metabolism.
Hyperestrogenism, hyperestrogenic state, or estrogen excess, is a medical condition characterized by an excessive amount of estrogenic activity in the body.

Leydig cell hypoplasia (LCH), also known as Leydig cell agenesis, is a rare autosomal recessive genetic and endocrine syndrome affecting an estimated 1 in 1,000,000 genetic males. It is characterized by an inability of the body to respond to luteinizing hormone (LH), a gonadotropin which is normally responsible for signaling Leydig cells of the testicles to produce testosterone and other androgen sex hormones. The condition manifests itself as pseudohermaphroditism, hypergonadotropic hypogonadism, reduced or absent puberty, and infertility.
Gonadotropin-releasing hormone (GnRH) insensitivity also known as Isolated gonadotropin-releasing hormone (GnRH)deficiency (IGD) is a rare autosomal recessive genetic and endocrine syndrome which is characterized by inactivating mutations of the gonadotropin-releasing hormone receptor (GnRHR) and thus an insensitivity of the receptor to gonadotropin-releasing hormone (GnRH), resulting in a partial or complete loss of the ability of the gonads to synthesize the sex hormones. The condition manifests itself as isolated hypogonadotropic hypogonadism (IHH), presenting with symptoms such as delayed, reduced, or absent puberty, low or complete lack of libido, and infertility, and is the predominant cause of IHH when it does not present alongside anosmia.
Follicle-stimulating hormone (FSH) insensitivity, or ovarian insensitivity to FSH in females, also referable to as ovarian follicle hypoplasia or granulosa cell hypoplasia in females, is a rare autosomal recessive genetic and endocrine syndrome affecting both females and males, with the former presenting with much greater severity of symptomatology. It is characterized by a resistance or complete insensitivity to the effects of follicle-stimulating hormone (FSH), a gonadotropin which is normally responsible for the stimulation of estrogen production by the ovaries in females and maintenance of fertility in both sexes. The condition manifests itself as hypergonadotropic hypogonadism, reduced or absent puberty, amenorrhea, and infertility in females, whereas males present merely with varying degrees of infertility and associated symptoms.
The fertile eunuch syndrome or Pasqualini syndrome is a cause of hypogonadotropic hypogonadism caused by a luteinizing hormone deficiency. It is characterized by hypogonadism with spermatogenesis. Pasqualini and Bur published the first case of eunuchoidism with preserved spermatogenesis in 1950 in la Revista de la Asociación Médica Argentina. The hypoandrogenism with spermatogenesis syndrome included:
Hypergonadotropic hypergonadism is an endocrine situation and subtype of hypergonadism in which both gonadotropin levels and gonadal function, such as sex hormone production, are abnormally high. It can be associated with hyperandrogenism and hyperestrogenism and with gonadal cysts and tumors. It can be caused by medications such as gonadotropins, gonadotropin-releasing hormone agonists, nonsteroidal antiandrogens, and selective estrogen receptor modulators, as well as conditions like human chorionic gonadotropin-secreting tumors, complete androgen insensitivity syndrome, and estrogen insensitivity syndrome.
| | This genetic disorder article is a stub. You can help Wikipedia by expanding it. |