A brain tumor occurs when abnormal cells form within the brain. There are two main types of tumors: malignant tumors and benign (non-cancerous) tumors. These can be further classified as primary tumors, which start within the brain, and secondary tumors, which most commonly have spread from tumors located outside the brain, known as brain metastasis tumors. All types of brain tumors may produce symptoms that vary depending on the size of the tumor and the part of the brain that is involved. Where symptoms exist, they may include headaches, seizures, problems with vision, vomiting and mental changes. Other symptoms may include difficulty walking, speaking, with sensations, or unconsciousness.

Tuberous sclerosis complex (TSC) is a rare multisystem autosomal dominant genetic disease that causes non-cancerous tumours to grow in the brain and on other vital organs such as the kidneys, heart, liver, eyes, lungs and skin. A combination of symptoms may include seizures, intellectual disability, developmental delay, behavioral problems, skin abnormalities, lung disease, and kidney disease.

Focal cortical dysplasia (FCD) is a congenital abnormality of brain development where the neurons in an area of the brain failed to migrate in the proper formation in utero. Focal means that it is limited to a focal zone in any lobe. Focal cortical dysplasia is a common cause of intractable epilepsy in children and is a frequent cause of epilepsy in adults. There are three types of FCD with subtypes, including type 1a, 1b, 1c, 2a, 2b, 3a, 3b, 3c, and 3d, each with distinct histopathological features. All forms of focal cortical dysplasia lead to disorganization of the normal structure of the cerebral cortex :

Neurofibromatosis type I (NF-1), or von Recklinghausen syndrome, is a complex multi-system human disorder caused by the mutation of Neurofibromin 1, a gene on chromosome 17 that is responsible for production of a protein (neurofibromin) which is needed for normal function in many human cell types. NF-1 causes tumors along the nervous system which can grow anywhere on the body. NF-1 is one of the most common genetic disorders and is not limited to any person's race or sex. NF-1 is an autosomal dominant disorder, which means that mutation or deletion of one copy of the NF-1 gene is sufficient for the development of NF-1, although presentation varies widely and is often different even between relatives affected by NF-1.

A hamartoma is a mostly benign, local malformation of cells that resembles a neoplasm of local tissue but is usually due to an overgrowth of multiple aberrant cells, with a basis in a systemic genetic condition, rather than a growth descended from a single mutated cell (monoclonality), as would typically define a benign neoplasm/tumor. Despite this, many hamartomas are found to have clonal chromosomal aberrations that are acquired through somatic mutations, and on this basis the term hamartoma is sometimes considered synonymous with neoplasm. Hamartomas are by definition benign, slow-growing or self-limiting, though the underlying condition may still predispose the individual towards malignancies.

Oligoastrocytomas are a subset of brain tumors that present with an appearance of mixed glial cell origin, astrocytoma and oligodendroglioma. However, the term "Oligoastrocytoma" is now considered obsolete by the National Comprehensive Cancer Network stating "the term should no longer be used as such morphologically ambiguous tumors can be reliably resolved into astrocytomas and oligodendrogliomas with molecular testing."

Gray matter heterotopia is a neurological disorder caused by gray matter being located in an atypical location in the brain.
Neuroepithelial cells, or neuroectodermal cells, form the wall of the closed neural tube in early embryonic development. The neuroepithelial cells span the thickness of the tube's wall, connecting with the pial surface and with the ventricular or lumenal surface. They are joined at the lumen of the tube by junctional complexes, where they form a pseudostratified layer of epithelium called neuroepithelium.

Choroid plexus papilloma, also known as papilloma of the choroid plexus, is a rare benign neuroepithelial intraventricular WHO grade I lesion found in the choroid plexus. It leads to increased cerebrospinal fluid production, thus causing increased intracranial pressure and hydrocephalus.

Medulloepithelioma is a rare, primitive, fast-growing brain tumour thought to stem from cells of the embryonic medullary cavity. Tumours originating in the ciliary body of the eye are referred to as embryonal medulloepitheliomas, or diktyomas.
Neuro-oncology is the study of brain and spinal cord neoplasms, many of which are very dangerous and life-threatening. Among the malignant brain cancers, gliomas of the brainstem and pons, glioblastoma multiforme, and high-grade astrocytoma/oligodendroglioma are among the worst. In these cases, untreated survival usually amounts to only a few months, and survival with current radiation and chemotherapy treatments may extend that time from around a year to a year and a half, possibly two or more, depending on the patient's condition, immune function, treatments used, and the specific type of malignant brain neoplasm. Surgery may in some cases be curative, but, as a general rule, malignant brain cancers tend to regenerate and emerge from remission easily, especially highly malignant cases. In such cases, the goal is to excise as much of the mass and as much of the tumor margin as possible without endangering vital functions or other important cognitive abilities. The Journal of Neuro-Oncology is the longest continuously published journal in the field and serves as a leading reference to those practicing in the area of neuro-oncology.
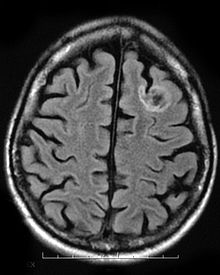
Fibrillary astrocytomas are a group of primary slow-growing brain tumors that typically occur in adults between the ages of 20 and 50.

Tuber cinereum hamartoma is a benign tumor in which a disorganized collection of neurons and glia accumulate at the tuber cinereum of the hypothalamus on the floor of the third ventricle. It is a congenital malformation, included on the spectrum of gray matter heterotopias. Formation occurs during embryogenesis, typically between days 33 and 41 of gestation. Size of the tumor varies from one to three centimeters in diameter, with the mean being closer to the low end of this range. It is estimated to occur at a frequency of one in one million individuals.

Subependymal giant cell astrocytoma is a low-grade astrocytic brain tumor (astrocytoma) that arises within the ventricles of the brain. It is most commonly associated with tuberous sclerosis complex (TSC). Although it is a low-grade tumor, its location can potentially obstruct the ventricles and lead to hydrocephalus.

Astroblastoma is a rare glial tumor derived from the astroblast, a type of cell that closely resembles spongioblastoma and astrocytes. Astroblastoma cells are most likely found in the supratentorial region of the brain that houses the cerebrum, an area responsible for all voluntary movements in the body. It also occurs significantly in the frontal lobe, parietal lobe, and temporal lobe, areas where movement, language creation, memory perception, and environmental surroundings are expressed. These tumors can be present in major brain areas not associated with the main cerebral hemispheres, including the cerebellum, optic nerve, cauda equina, hypothalamus, and brain stem.
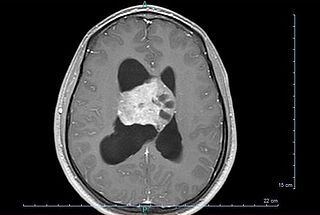
Central neurocytoma (CNC) is an extremely rare, ordinarily benign intraventricular brain tumour that typically forms from the neuronal cells of the septum pellucidum. The majority of central neurocytomas grow inwards into the ventricular system forming interventricular neurocytomas. This leads to two primary symptoms of CNCs, blurred vision and increased intracranial pressure. Treatment for a central neurocytoma typically involves surgical removal, with an approximate 1 in 5 chance of recurrence. Central neurocytomas are classified as a grade II tumor under the World Health Organization's classification of tumors of the nervous system.

Ulegyria is a diagnosis used to describe a specific type of cortical scarring in the deep regions of the sulcus that leads to distortion of the gyri. Ulegyria is identified by its characteristic "mushroom-shaped" gyri, in which scarring causes shrinkage and atrophy in the deep sulcal regions while the surface gyri are spared. This condition is most often caused by hypoxic-ischemic brain injury in the perinatal period. The effects of ulegyria can range in severity, although it is most commonly associated with cerebral palsy, mental retardation and epilepsy. N.C. Bresler was the first to view ulegyria in 1899 and described this abnormal morphology in the brain as “mushroom-gyri." Although ulegyria was first identified in 1899, there is still limited information known or reported about the condition.

James Rutka is a Canadian neurosurgeon from Toronto, Canada. Rutka served as RS McLaughlin Professor and Chair of the Department of Surgery in the Faculty of Medicine at the University of Toronto from 2011 – 2022. He subspecializes in pediatric neurosurgery at The Hospital for Sick Children (SickKids), and is a Senior Scientist in the Research Institute at SickKids. His main clinical interests include the neurosurgical treatment of children with brain tumours and epilepsy. His research interests lie in the molecular biology of human brain tumours – specifically in the determination of the mechanisms by which brain tumours grow and invade. He is the Director of the Arthur and Sonia Labatt Brain Tumour Research Centre at SickKids, and Editor-in-Chief of the Journal of Neurosurgery.

Occipital epilepsy is a neurological disorder that arises from excessive neural activity in the occipital lobe of the brain that may or may not be symptomatic. Occipital lobe epilepsy is fairly rare, and may sometimes be misdiagnosed as migraine when symptomatic. Epileptic seizures are the result of synchronized neural activity that is excessive, and may stem from a failure of inhibitory neurons to regulate properly.

Angiocentric glioma (AG) refers to a rare neuroepithelial tumor when the superficial brain malignant cells enclose the brain vessels, commonly found in children and young adults. Initially identified in 2005 by Wang and his team from the University of Texas, AG was classified as Grade I by 2007 WHO Classification of Tumors of the Central Nervous System due to its benign clinical behavior, low proliferation index, and curative properties. AG primarily affects children and young adults at an average initial diagnosis age of 16 years old. Over 85% AG patients experience intractable seizures since childhood, especially partial epilepsy.