Neuroblastoma (NB) is a type of cancer that forms in certain types of nerve tissue.[1] It most frequently starts from one of the adrenal glands but can also develop in the head, neck, chest, abdomen, or spine.[1] Symptoms may include bone pain, a lump in the abdomen, neck, or chest, or a painless bluish lump under the skin.[1]
Typically, neuroblastoma occurs due to a genetic mutation occurring in the first trimester of pregnancy.[4][5] Rarely, it may be due to a mutation inherited.[1] Environmental factors have not been found to be involved.[2] Diagnosis is based on a tissue biopsy.[1] Occasionally, it may be found in a baby by ultrasound during pregnancy.[1] At diagnosis, the cancer has usually already spread.[1] The cancer is divided into low-, intermediate-, and high-risk groups based on a child's age, cancer stage, and what the cancer looks like.[1]
Treatment and outcomes depends on the risk group a person is in.[1][5] Treatments may include observation, surgery, radiation, chemotherapy, or stem cell transplantation.[1] Low-risk disease in babies typically has a good outcome with surgery or simply observation.[5] In high-risk disease, chances of long-term survival, however, are less than 40%, despite aggressive treatment.[5]
Neuroblastoma is the most common cancer in babies and the third-most common cancer in children after leukemia and brain cancer.[5] About one in every 7,000 children is affected at some time.[2] About 90% of cases occur in children less than 5 years old, and it is rare in adults.[2][3] Of cancer deaths in children, about 15% are due to neuroblastoma.[3] The disease was first described in the 1800s.[6]
Signs and symptoms
The first symptoms of neuroblastoma are often vague, making diagnosis difficult. Fatigue, loss of appetite, fever, and joint pain are common. Symptoms depend on primary tumor locations and metastases if present:[7]
A tumor pressing on the spinal cord may cause weakness, thus an inability to stand, crawl, or walk.
Bone lesions in the legs and hips may cause pain and limping.
A tumor in the bones around the eyes or orbits may cause distinct bruising and swelling.
Infiltration of the bone marrow may cause pallor from anemia.
Neuroblastoma often spreads to other parts of the body before any symptoms are apparent, and 50 to 60% of all neuroblastoma cases present with metastases.[8]
The most common location for neuroblastoma to originate (i.e., the primary tumor) is in the adrenal glands. This occurs in 40% of localized tumors and in 60% of cases of widespread disease. Neuroblastoma can also develop anywhere along the sympathetic nervous system chain from the neck to the pelvis. Frequencies in different locations include: neck (1%), chest (19%), abdomen (30% nonadrenal), or pelvis (1%). In rare cases, no primary tumor can be discerned.[9]
The cause of neuroblastoma is not well understood. The great majority of cases are sporadic and nonfamilial. About 1–2% of cases run in families and have been linked to specific gene mutations. Familial neuroblastoma in some cases is caused by rare germline mutations in the anaplastic lymphoma kinase (ALK) gene.[12] Germline mutations in the PHOX2Bor KIF1B gene have been implicated in familial neuroblastoma, as well. Neuroblastoma is also a feature of neurofibromatosis type 1 and the Beckwith-Wiedemann syndrome.
MYCNoncogene amplification within the tumor is a common finding in neuroblastoma. The degree of amplification shows a bimodal distribution: either 3- to 10-fold, or 100- to 300-fold. The presence of this mutation is highly correlated to advanced stages of disease.[13]
Duplicated segments of the LMO1 gene within neuroblastoma tumor cells have been shown to increase the risk of developing an aggressive form of the cancer.[14]
Other genes might have a prognostic role in neuroblastoma. A bioinformatics study published in 2023 suggested that the AHCY, DPYSL3, and NME1genes might have a prognostic role in this disease.[15]
One study strongly indicates that miRNAs that are excessively expressed in 1p-deleted neuroblastoma cells, as opposed to other genetic subgroups of neuroblastoma, could potentially disrupt the regulation of genes associated with neuronal differentiation, thereby contribute to the pathogenesis of neuroblastoma. Furthermore, it was noted that miR-495 primarily targeted the majority of mRNAs that are involved in neuronal differentiation.[17]
Several risk factors have been proposed and are the subject of ongoing research. Due to characteristic early onset, many studies have focused on parental factors around conception and during gestation. Factors investigated have included occupation (i.e. exposure to chemicals in specific industries), smoking, alcohol consumption, use of medicinal drugs during pregnancy, and birth factors; however, results have been inconclusive.[18]
Other studies have examined possible links with atopy and exposure to infection early in life,[19] use of hormones and fertility drugs,[20] and maternal use of hair dye.[21][22]
Diagnosis
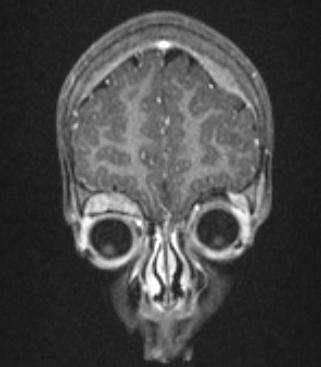
MRI showing orbital and skull vault metastatic NB in 2-year-old
The diagnosis is usually confirmed by a surgical pathologist, taking into account the clinical presentation, microscopic findings, and other laboratory tests. It may arise from any neural crest element of the sympathetic nervous system (SNS).
Esthesioneuroblastoma, also known as olfactory neuroblastoma, is believed to arise from the olfactory epithelium and its classification remains controversial. However, since it is not a sympathetic nervous system malignancy, esthesioneuroblastoma is a distinct clinical entity and is not to be confused with neuroblastoma.[23][24]
Another way to detect neuroblastoma is the meta-iodobenzylguanidine scan, which is taken up by 90 to 95% of all neuroblastomas, often termed "mIBG-avid".[26] The mechanism is that mIBG is taken up by sympathetic neurons, and is a functioning analog of the neurotransmitter norepinephrine. When it is radio-iodinated with I-131 or I-123 (radioactive iodine isotopes), it is a very good radiopharmaceutical for diagnosis and monitoring of response to treatment for this disease. With a half-life of 13 hours, I-123 is the preferred isotope for imaging sensitivity and quality. I-131 has a half-life of 8 days and at higher doses is an effective therapy as targeted radiation against relapsed and refractory neuroblastoma.[27] As mIBG is not always taken up by neuroblastomas, researchers have explored in children with neuroblastoma whether another type of nuclear imaging, fluoro-deoxy-glucose – positron emission tomography, often termed "F-FDG-PET", might be useful.[28] Evidence suggests that this might be advisable to use in children with neuroblastoma for which mIBG does not work, but more research is needed in this area.[28]
Histology
Microscopic view of stroma-rich ganglioneuroblastoma
On microscopy, the tumor cells are typically described as small, round and blue, and rosette patterns (Homer Wright pseudorosettes) may be seen. Homer Wright pseudorosettes are tumor cells around the neuropil, not to be confused with a true rosettes, which are tumor cells around an empty lumen.[29] They are also distinct from the pseudorosettes of an ependymoma which consist of tumor cells with glial fibrillary acidic protein (GFAP)–positive processes tapering off toward a blood vessel (thus a combination of the two).[30] A variety of immunohistochemical stains are used by pathologists to distinguish neuroblastomas from histological mimics, such as rhabdomyosarcoma, Ewing's sarcoma, lymphoma and Wilms' tumor.[31]
Neuroblastoma is one of the peripheral neuroblastic tumors (pNTs) that have similar origins and show a wide pattern of differentiation ranging from benignganglioneuroma to stroma-rich ganglioneuroblastoma with neuroblastic cells intermixed or in nodules, to highly malignant neuroblastoma. This distinction in the pre-treatment tumor pathology is an important prognostic factor, along with age and mitosis-karyorrhexis index (MKI). This pathology classification system (the Shimada system) describes "favorable" and "unfavorable" tumors by the International Neuroblastoma Pathology Committee (INPC) which was established in 1999 and revised in 2003.[32]
Staging
The "International Neuroblastoma Staging System" (INSS) established in 1986 and revised in 1988 stratifies neuroblastoma according to its anatomical presence at diagnosis:[33][34][35]
Stage 1: Localized tumor confined to the area of origin.
Stage 2A: Unilateral tumor with incomplete gross resection; identifiable ipsilateral and contralateral lymph node negative for tumor.
Stage 2B: Unilateral tumor with complete or incomplete gross resection; with ipsilateral lymph node positive for tumor; identifiable contralateral lymph node negative for tumor.
Stage 3: Tumor infiltrating across midline with or without regional lymph node involvement; or unilateral tumor with contralateral lymph node involvement; or midline tumor with bilateral lymph node involvement.
Stage 4: Dissemination of tumor to distant lymph nodes, bone marrow, bone, liver, or other organs except as defined by Stage 4S.
Stage 4S: Age <1 year old with localized primary tumor as defined in Stage 1 or 2, with dissemination limited to liver, skin, or bone marrow (less than 10 percent of nucleated bone marrow cells are tumors).
Although international agreement on staging (INSS) has been used, the need for an international consensus on risk assignment has also been recognized in order to compare similar cohorts in results of studies. Beginning in 2005, representatives of the major pediatric oncology cooperative groups have met to review data for 8,800 people with neuroblastoma treated in Europe, Japan, USA, Canada, and Australia between 1990 and 2002. This task force has proposed the International Neuroblastoma Risk Group (INRG) classification system. Retrospective studies revealed the high survival rate of 12–18 month-old age group, previously categorized as high-risk, and prompted the decision to reclassify 12–18 month-old children without N-myc (also commonly referred to as MYCN) amplification to intermediate risk category.[36]
The new INRG risk assignment will classify neuroblastoma at diagnosis based on a new International Neuroblastoma Risk Group Staging System (INRGSS):
Stage L1: Localized disease without image-defined risk factors.
Stage L2: Localized disease with image-defined risk factors.
Stage M: Metastatic disease.
Stage MS: Metastatic disease "special" where MS is equivalent to stage 4S.
The new risk stratification will be based on the new INRGSS staging system, age (dichotomized at 18 months), tumor grade, N-myc amplification, unbalanced 11q aberration, and ploidy into four pre-treatment risk groups: very low, low, intermediate, and high risk.[5][37]
Screening
Urine catecholamine level can be elevated in pre-clinical neuroblastoma. Screening asymptomatic infants at three weeks, six months, and one year has been performed in Japan, Canada, Austria and Germany since the 1980s.[38][39] Japan began screening six-month-olds for neuroblastoma via analysis of the levels of homovanillic acid and vanilmandelic acid in 1984. Screening was halted in 2004 after studies in Canada and Germany showed no reduction in deaths due to neuroblastoma, but rather caused an increase in diagnoses that would have disappeared without treatment, subjecting those infants to unnecessary surgery and chemotherapy.[40][41][42]
Biologic and genetic characteristics have been identified, which, when added to classic clinical staging, has allowed assignment to risk groups for planning treatment intensity.[44] These criteria include the age of the person, extent of disease spread, microscopic appearance, and genetic features including DNA ploidy and N-myconcogene amplification (N-myc regulates microRNAs[45]), into low, intermediate, and high risk disease. A recent biology study (COG ANBL00B1) analyzed 2687 people with neuroblastoma and the spectrum of risk assignment was determined: 37% of neuroblastoma cases are low risk, 18% are intermediate risk, and 45% are high risk.[46] (There is some evidence that the high- and low-risk types are caused by different mechanisms, and are not merely two different degrees of expression of the same mechanism.)[47]
The therapies for these different risk categories are very different.
Intermediate-risk disease is treated with surgery and chemotherapy.[49]
High-risk neuroblastoma is treated with intensive chemotherapy, surgery, radiation therapy, bone marrow / hematopoietic stem cell transplantation,[50] biological-based therapy with 13-cis-retinoic acid (isotretinoin or Accutane)[51] and antibody therapy usually administered with the cytokinesGM-CSF and IL-2.[52] A meta analysis has found evidence that in children with high-risk neuroblastoma, treatment with myeloablative therapy improves event-free survival but may increase the risk of side effects such as kidney problems when compared to conventional chemotherapy.[53]
People with low and intermediate risk disease have an excellent prognosis with cure rates above 90% for low risk and 70–90% for intermediate risk. In contrast, therapy for high-risk neuroblastoma the past two decades[when?] resulted in cures only about 30% of the time.[54] The addition of antibody therapy has raised survival rates for high-risk disease significantly. In March 2009, an early analysis of a Children's Oncology Group (COG) study with 226 people that are high-risk showed that two years after stem cell transplant 66% of the group randomized to receive ch14.18 antibody with GM-CSF and IL-2 were alive and disease-free compared to only 46% in the group that did not receive the antibody. The randomization was stopped so all people enrolling on the trial would receive the antibody therapy.[55]
Chemotherapy agents used in combination have been found to be effective against neuroblastoma. Agents commonly used in induction and for stem cell transplant conditioning are platinum compounds (cisplatin, carboplatin), alkylating agents (cyclophosphamide, ifosfamide, melphalan), topoisomerase II inhibitor (etoposide), anthracycline antibiotics (doxorubicin) and vinca alkaloids (vincristine). Some newer regimens include topoisomerase I inhibitors (topotecan and irinotecan) in induction which have been found to be effective against recurrent disease. Although further research is needed, interventions currently under pre-clinical investigation include epigenetic therapies, such as inhibition of SWI/SNF,[56] which may complement existing retinoid therapies.
In November 2020, naxitamab was approved for medical use in the United States in combination with granulocyte-macrophage colony-stimulating factor (GM-CSF) to treat people one year of age and older with high-risk neuroblastoma in bone or bone marrow whose tumor did not respond to or has come back after previous treatments and has shown a partial response, minor response, or stable disease to prior therapy.[57][58]
Prognosis
By data from England, the overall 5-year survival rate of neuroblastoma is 67%.[59] Between 20% and 50% of high-risk cases do not respond adequately to induction high-dose chemotherapy and are progressive or refractory.[60][61] Relapse after completion of frontline therapy is also common. Further treatment is available in phase I and phase II clinical trials that test new agents and combinations of agents against neuroblastoma, but the outcome remains very poor for relapsed high-risk disease.[62]
Most long-term survivors alive today had low or intermediate risk disease and milder courses of treatment compared to high-risk disease. The majority of survivors have long-term effects from the treatment. Survivors of intermediate and high-risk treatment often experience hearing loss, growth reduction, thyroid function disorders, learning difficulties, and greater risk of secondary cancers affect survivors of high-risk disease.[63][64] An estimated two of three survivors of childhood cancer will ultimately develop at least one chronic and sometimes life-threatening health problem within 20 to 30 years after the cancer diagnosis.[65][66][67]
Cytogenetic profiles
Based on a series of 493 neuroblastoma samples, it has been reported that overall genomic pattern, as tested by array-based karyotyping, is a predictor of outcome in neuroblastoma:[68]
Tumors presenting exclusively with whole chromosome copy number changes were associated with excellent survival.
Tumors presenting with any kind of segmental chromosome copy number changes were associated with a high risk of relapse.
Within tumors showing segmental alterations, additional independent predictors of decreased overall survival were N-myc amplification, 1p and 11q deletions, and 1q gain.
Earlier publications categorized neuroblastomas into three major subtypes based on cytogenetic profiles:[69][70]
Subtype 1: favorable neuroblastoma with near triploidy and a predominance of numerical gains and losses, mostly representing non-metastatic NB stages 1, 2 and 4S.
Subtypes 2A and 2B: found in unfavorable widespread neuroblastoma, stages 3 and 4, with 11q loss and 17q gain without N-myc amplification (subtype 2A) or with N-myc amplification often together with 1p deletions and 17q gain (subtype 2B).
Virtual karyotyping can be performed on fresh or paraffin-embedded tumors to assess copy number at these loci. SNP array virtual karyotyping is preferred for tumor samples, including neuroblastomas, because they can detect copy neutral loss of heterozygosity (acquired uniparental disomy). Copy neutral LOH can be biologically equivalent to a deletion and has been detected at key loci in neuroblastoma.[71] ArrayCGH, FISH, or conventional cytogenetics cannot detect copy neutral LOH.
Epidemiology
Incidences and prognoses of adrenal tumors, with "neuronal tumor" at right
Neuroblastoma comprises 6–10% of all childhood cancers, and 15% of cancer deaths in children. The annual mortality rate is 10 per million children in the 0- to 4-year-old age group, and 4 per million in the 4- to 9-year old age group.[73]
The highest number of cases is in the first year of life, and some cases are congenital. The age range is broad, including older children and adults,[74] but only 10% of cases occur in people older than 5 years of age.[26] A large European study reported less than 2% of over 4000 neuroblastoma cases were over 18 years old.[75]
History
Rudolf Virchow: the first to describe an abdominal tumor in a child as a "glioma"
In 1864 German physician Rudolf Virchow was the first to describe an abdominal tumor in a child as a "glioma". The characteristics of tumors from the sympathetic nervous system and the adrenal medulla were then noted in 1891 by German pathologist Felix Marchand.[76][77] In 1901 the distinctive presentation of stage 4S in infants (liver but no bone metastases) was described by William Pepper. In 1910 James Homer Wright understood the tumor to originate from primitive neural cells, and named it neuroblastoma. He also noted the circular clumps of cells in bone marrow samples which are now termed "Homer Wright rosettes". Of note, "Homer-Wright" with a hyphen is grammatically incorrect, as the eponym refers to just Dr. Wright.[78]
Scientific research
Microscopic view of a NB cell line (SH-SY5Y) used in preclinical research for testing new agents
Preclinical models
Neuroblastoma patient derived tumor xenografts (PDXs) have been created by orthotopic implantation of tumor samples into immunodeficient mice.[79] PDX models have several advantages over conventional cancer cell lines (CCL)s.[80] Neuroblastoma PDXs retain the genetic hallmarks of their corresponding tumors and PDXs display infiltrative growth and metastasis to distant organs.[79] PDX models are more predictive of clinical outcome as compared to conventional cancer cell line xenografts.[81] Neuroblastoma PDXs might thus serve as clinically relevant models to identify effective compounds against neuroblastoma.[79]
Treatments
Recent focus has been to reduce therapy for low and intermediate risk neuroblastoma while maintaining survival rates at 90%.[82] A study of 467 people that are at intermediate risk enrolled in A3961 from 1997 to 2005 confirmed the hypothesis that therapy could be successfully reduced for this risk group. Those with favorable characteristics (tumor grade and response) received four cycles of chemotherapy, and those with unfavorable characteristics received eight cycles, with three-year event free survival and overall survival stable at 90% for the entire cohort. Future plans are to intensify treatment for those people with aberration of 1p36 or 11q23 chromosomes as well as for those who lack early response to treatment.[83][84]
By contrast, focus the past 20 years or more has been to intensify treatment for high-risk neuroblastoma. Chemotherapy induction variations, timing of surgery, stem cell transplant regimens, various delivery schemes for radiation, and use of monoclonal antibodies and retinoids to treat minimal residual disease continue to be examined. Recent phase III clinical trials with randomization have been carried out to answer these questions to improve survival of high-risk disease:
Refractory and relapsed neuroblastoma
Chemotherapy with topotecan and cyclophosphamide is frequently used in refractory setting and after relapse.[85]
A haploidentical stem cell transplant, that is, donor cells derived from parents, is being studied in those with refractory or relapsing neuroblastoma as stem cells from the person themselves is not useful.[86]
It has been shown that neuroblastoma display a high expression of somatostatin receptors[87][88][89] and this enables potential therapy using 177Lu-DOTA-TATE, a type of radionuclide therapy that specifically targets the somatostatin receptors. Several early phase clinical trials using 177Lu-DOTA-TATE for treatment of high-risk refractory/relapsed neuroblastoma have been conducted with promising results.[90][91][92]
Electronic health records' data
Several international initiatives have been recently launched for the sharing of data of electronic health records of patients with neuroblastoma: these data in fact can be analyzed with machine learning and statistics models to infer new knowledge about this disease. To this end, the International Neuroblastoma Risk Group (INRG) recently released the INRG Data Commons,[93] while University of Chicago launched the Pediatric Cancer Data Commons.[94] These two repositories contain data of electronic health records of thousands of patients that are available for scientific research, with prior authorization. In 2022, researchers released a new data repository of electronic health records called Neuroblastoma Electronic Health Records Open Data Repository where data can be downloaded freely without any restriction.[95]
Organisations
The Advances in Neuroblastoma Research Association (ANRA) is the peak body for researchers in neuroblastoma biology, diagnosis, prognosis, and therapy, and conducts meetings every two years to exchange information among them.[96]
↑ Olshan AF, Bunin GR (2000). "Epidemiology of Neuroblastoma". In Brodeur GM, Sawada T, Tsuchida Y, Voûte PP (eds.). Neuroblastoma. Amsterdam: Elsevier. pp.33–9. ISBN978-0-444-50222-3.
↑ Heck JE, Ritz B, Hung RJ, Hashibe M, Boffetta P (March 2009). "The epidemiology of neuroblastoma: a review". Paediatric and Perinatal Epidemiology. 23 (2): 125–143. doi:10.1111/j.1365-3016.2008.00983.x. PMID19159399.
↑ Carter RL, al-Sams SZ, Corbett RP, Clinton S (May 1990). "A comparative study of immunohistochemical staining for neuron-specific enolase, protein gene product 9.5 and S-100 protein in neuroblastoma, Ewing's sarcoma and other round cell tumours in children". Histopathology. 16 (5): 461–467. doi:10.1111/j.1365-2559.1990.tb01545.x. PMID2163356. S2CID6461880.
↑ Brodeur GM, Seeger RC, Barrett A, Berthold F, Castleberry RP, D'Angio G, etal. (December 1988). "International criteria for diagnosis, staging, and response to treatment in patients with neuroblastoma". Journal of Clinical Oncology. 6 (12): 1874–1881. doi:10.1200/JCO.1988.6.12.1874. PMID3199170.
↑ Brodeur GM, Pritchard J, Berthold F, Carlsen NL, Castel V, Castelberry RP, etal. (August 1993). "Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment". Journal of Clinical Oncology. 11 (8): 1466–1477. doi:10.1200/JCO.1993.11.8.1466. PMID8336186.
↑ Schilling FH, Spix C, Berthold F, Erttmann R, Sander J, Treuner J, Michaelis J (July 2003). "Children may not benefit from neuroblastoma screening at 1 year of age. Updated results of the population based controlled trial in Germany". Cancer Letters. 197 (1–2): 19–28. doi:10.1016/S0304-3835(03)00077-6. PMID12880955.
↑ Kushner BH, Kramer K, LaQuaglia MP, Modak S, Yataghene K, Cheung NK (December 2004). "Reduction from seven to five cycles of intensive induction chemotherapy in children with high-risk neuroblastoma". Journal of Clinical Oncology. 22 (24): 4888–4892. doi:10.1200/JCO.2004.02.101. PMID15611504.
↑ Ceschel S, Casotto V, Valsecchi MG, Tamaro P, Jankovic M, Hanau G, etal. (October 2006). "Survival after relapse in children with solid tumors: a follow-up study from the Italian off-therapy registry". Pediatric Blood & Cancer. 47 (5): 560–566. doi:10.1002/pbc.20726. PMID16395684. S2CID31490896.
↑ Gurney JG, Tersak JM, Ness KK, Landier W, Matthay KK, Schmidt ML (November 2007). "Hearing loss, quality of life, and academic problems in long-term neuroblastoma survivors: a report from the Children's Oncology Group". Pediatrics. 120 (5): e1229 –e1236. doi:10.1542/peds.2007-0178. PMID17974716. S2CID10606999.
↑ Michels E, Vandesompele J, Hoebeeck J, Menten B, De Preter K, Laureys G, etal. (2006). "Genome wide measurement of DNA copy number changes in neuroblastoma: dissecting amplicons and mapping losses, gains and breakpoints". Cytogenetic and Genome Research. 115 (3–4): 273–282. doi:10.1159/000095924. PMID17124410. S2CID14012430.
↑ Brodeur GM, Hogarty MD, Mosse YP, Maris JM (1997). "Neuroblastoma". In Pizzo PA, Poplack DG (eds.). Principles and Practice of Pediatric Oncology (6thed.). Wolters Kluwer Health/Lippincott Williams & Wilkins. pp.886–922. ISBN978-1-60547-682-7.
↑ Rothenberg AB, Berdon WE, D'Angio GJ, Yamashiro DJ, Cowles RA (February 2009). "Neuroblastoma-remembering the three physicians who described it a century ago: James Homer Wright, William Pepper, and Robert Hutchison". Pediatric Radiology. 39 (2): 155–160. doi:10.1007/s00247-008-1062-z. PMID19034443. S2CID19611725.
↑ Morgenstern DA, Baruchel S, Irwin MS (July 2013). "Current and future strategies for relapsed neuroblastoma: challenges on the road to precision therapy". Journal of Pediatric Hematology/Oncology. 35 (5): 337–347. doi:10.1097/MPH.0b013e318299d637. PMID23703550. S2CID5529288.
↑ Chicco, D., Cerono, G., Cangelosi, D. (2022), "A survey on publicly available open datasets derived from electronic health records (EHRs) of patients with neuroblastoma", Data Science Journal, 21 (1): 17, doi:10.5334/dsj-2022-017, hdl:10281/430238
↑ "Home". Advances in Neuroblastoma Research Association. 26 April 2014. Retrieved 11 April 2024.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.





{kind=link}