Related Research Articles

A brain tumor occurs when abnormal cells form within the brain. There are two main types of tumors: malignant tumors and benign (non-cancerous) tumors. These can be further classified as primary tumors, which start within the brain, and secondary tumors, which most commonly have spread from tumors located outside the brain, known as brain metastasis tumors. All types of brain tumors may produce symptoms that vary depending on the size of the tumor and the part of the brain that is involved. Where symptoms exist, they may include headaches, seizures, problems with vision, vomiting and mental changes. Other symptoms may include difficulty walking, speaking, with sensations, or unconsciousness.

An ependymoma is a tumor that arises from the ependyma, a tissue of the central nervous system. Usually, in pediatric cases the location is intracranial, while in adults it is spinal. The common location of intracranial ependymomas is the fourth ventricle. Rarely, ependymomas can occur in the pelvic cavity.

Glioblastoma, previously known as glioblastoma multiforme (GBM), is one of the most aggressive types of cancer that begin within the brain. Initially, signs and symptoms of glioblastoma are nonspecific. They may include headaches, personality changes, nausea, and symptoms similar to those of a stroke. Symptoms often worsen rapidly and may progress to unconsciousness.
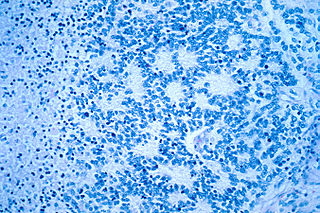
Neuroblastoma (NB) is a type of cancer that forms in certain types of nerve tissue. It most frequently starts from one of the adrenal glands but can also develop in the neck, chest, abdomen, or spine. Symptoms may include bone pain, a lump in the abdomen, neck, or chest, or a painless bluish lump under the skin.

Desmoplastic small-round-cell tumor (DSRCT) is an aggressive and rare cancer that primarily occurs as masses in the abdomen. Other areas affected may include the lymph nodes, the lining of the abdomen, diaphragm, spleen, liver, chest wall, skull, spinal cord, large intestine, small intestine, bladder, brain, lungs, testicles, ovaries, and the pelvis. Reported sites of metastatic spread include the liver, lungs, lymph nodes, brain, skull, and bones. It is characterized by the EWS-WT1 fusion protein.
A blastoma is a type of cancer, more common in children, that is caused by malignancies in precursor cells, often called blasts. Examples are nephroblastoma, medulloblastoma, and retinoblastoma. The suffix -blastoma is used to imply a tumor of primitive, incompletely differentiated cells, e.g., chondroblastoma is composed of cells resembling the precursor of chondrocytes.

Medulloblastoma is a common type of primary brain cancer in children. It originates in the part of the brain that is towards the back and the bottom, on the floor of the skull, in the cerebellum, or posterior fossa.
Neuroepithelial cells, or neuroectodermal cells, form the wall of the closed neural tube in early embryonic development. The neuroepithelial cells span the thickness of the tube's wall, connecting with the pial surface and with the ventricular or lumenal surface. They are joined at the lumen of the tube by junctional complexes, where they form a pseudostratified layer of epithelium called neuroepithelium.

Primitive neuroectodermal tumor is a malignant (cancerous) neural crest tumor. It is a rare tumor, usually occurring in children and young adults under 25 years of age. The overall 5 year survival rate is about 53%.

An atypical teratoid rhabdoid tumor (AT/RT) is a rare tumor usually diagnosed in childhood. Although usually a brain tumor, AT/RT can occur anywhere in the central nervous system (CNS), including the spinal cord. About 60% will be in the posterior cranial fossa. One review estimated 52% in the posterior fossa, 39% are supratentorial primitive neuroectodermal tumors (sPNET), 5% are in the pineal, 2% are spinal, and 2% are multifocal.
Ectomesenchymoma is a rare, fast-growing tumor of the nervous system or soft tissue that occurs mainly in children, although cases have been reported in patients up to age 60. Ectomesenchymomas may form in the head and neck, abdomen, perineum, scrotum, or limbs. Also called malignant ectomesenchymoma.

A choroid plexus carcinoma is a type of choroid plexus tumor that affects the choroid plexus of the brain. It is considered the worst of the three grades of chord plexus tumors, having a much poorer prognosis than choroid atypical plexus papilloma and choroid plexus papilloma. The disease creates lesions in the brain and increases cerebrospinal fluid volume, resulting in hydrocephalus.

The following is a simplified (deprecated) version of the 2021 WHO classification of the tumours of the central nervous system. Currently, as of 2021, clinicians are using the WHO grade 5th edition, which incorporates recent advances in molecular pathology.

Medulloepithelioma is a rare, primitive, fast-growing brain tumour thought to stem from cells of the embryonic medullary cavity. Tumours originating in the ciliary body of the eye are referred to as embryonal medulloepitheliomas, or diktyomas.
Neuro-oncology is the study of brain and spinal cord neoplasms, many of which are very dangerous and life-threatening. Among the malignant brain cancers, gliomas of the brainstem and pons, glioblastoma multiforme, and high-grade astrocytoma/oligodendroglioma are among the worst. In these cases, untreated survival usually amounts to only a few months, and survival with current radiation and chemotherapy treatments may extend that time from around a year to a year and a half, possibly two or more, depending on the patient's condition, immune function, treatments used, and the specific type of malignant brain neoplasm. Surgery may in some cases be curative, but, as a general rule, malignant brain cancers tend to regenerate and emerge from remission easily, especially highly malignant cases. In such cases, the goal is to excise as much of the mass and as much of the tumor margin as possible without endangering vital functions or other important cognitive abilities. The Journal of Neuro-Oncology is the longest continuously published journal in the field and serves as a leading reference to those practicing in the area of neuro-oncology.
Pediatric ependymomas are similar in nature to the adult form of ependymoma in that they are thought to arise from radial glial cells lining the ventricular system. However, they differ from adult ependymomas in which genes and chromosomes are most often affected, the region of the brain they are most frequently found in, and the prognosis of the patients. Children with certain hereditary diseases, such as neurofibromatosis type II (NF2), have been found to be more frequently afflicted with this class of tumors, but a firm genetic link remains to be established. Symptoms associated with the development of pediatric ependymomas are varied, much like symptoms for a number of other pediatric brain tumors including vomiting, headache, irritability, lethargy, and changes in gait. Although younger children and children with invasive tumor types generally experience less favorable outcomes, total removal of the tumors is the most conspicuous prognostic factor for both survival and relapse.

Astroblastoma is a rare glial tumor derived from the astroblast, a type of cell that closely resembles spongioblastoma and astrocytes. Astroblastoma cells are most likely found in the supratentorial region of the brain that houses the cerebrum, an area responsible for all voluntary movements in the body. It also occurs significantly in the frontal lobe, parietal lobe, and temporal lobe, areas where movement, language creation, memory perception, and environmental surroundings are expressed. These tumors can be present in major brain areas not associated with the main cerebral hemispheres, including the cerebellum, optic nerve, cauda equina, hypothalamus, and brain stem.

A central nervous system primitive neuroectodermal tumor, often abbreviated as PNET, supratentorial PNET, or CNS-PNET, is one of the 3 types of embryonal central nervous system tumors defined by the World Health Organization. It is considered an embryonal tumor because it arises from cells partially differentiated or still undifferentiated from birth. Those cells are usually neuroepithelial cells, stem cells destined to turn into glia or neurons. It can occur anywhere within the spinal cord and cerebrum and can have multiple sites of origins, with a high probability of metastasis through cerebrospinal fluid (CSF).

In histopathology, a palisade is a single layer of relatively long cells, arranged loosely perpendicular to a surface and parallel to each other. A rosette is a palisade in a halo or spoke-and-wheel arrangement, surrounding a central core or hub. A pseudorosette is a perivascular radial arrangement of neoplastic cells around a small blood vessel.

Angiocentric glioma (AG) refers to a rare neuroepithelial tumor when the superficial brain malignant cells enclose the brain vessels, commonly found in children and young adults. Initially identified in 2005 by Wang and his team from the University of Texas, AG was classified as Grade I by 2007 WHO Classification of Tumors of the Central Nervous System due to its benign clinical behavior, low proliferation index, and curative properties. AG primarily affects children and young adults at an average initial diagnosis age of 16 years old. Over 85% AG patients experience intractable seizures since childhood, especially partial epilepsy.
References
- 1 2 Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM, Reifenberger G, Soffietti R, von Deimling A, Ellison DW (Aug 2021). "The 2021 WHO Classification of Tumors of the Central Nervous System: a summary". Neuro-oncology. 23 (8): 1231–1251. doi:10.1093/neuonc/noab106. PMC 7423804 . PMID 32601913.
- 1 2 Lambo S, von Hoff K, Korshunov A, Pfister SM, Kool M (Jun 2020). "ETMR: a tumor entity in its infancy". Acta Neuropathologica. 140 (3): 249–266. doi:10.1007/s00401-020-02182-2. PMC 8328013 . PMID 34185076.
- ↑ Korshunov A, Sturm D, Ryzhova M, Kool M (Dec 2013). "Embryonal tumor with abundant neuropil and true rosettes (ETANTR), ependymoblastoma, and medulloepithelioma share molecular similarity and comprise a single clinicopathological entity". Acta Neuropathologica. 128 (2): 279–289. doi:10.1007/s00401-013-1228-0. PMC 4102829 . PMID 24337497.