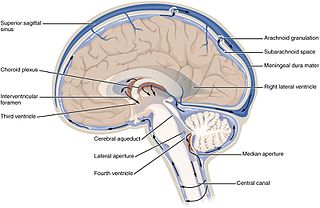
Cerebrospinal fluid (CSF) is a clear, colorless body fluid found within the tissue that surrounds the brain and spinal cord of all vertebrates.

A brain tumor occurs when abnormal cells form within the brain. There are two main types of tumors: malignant tumors and benign (non-cancerous) tumors. These can be further classified as primary tumors, which start within the brain, and secondary tumors, which most commonly have spread from tumors located outside the brain, known as brain metastasis tumors. All types of brain tumors may produce symptoms that vary depending on the size of the tumor and the part of the brain that is involved. Where symptoms exist, they may include headaches, seizures, problems with vision, vomiting and mental changes. Other symptoms may include difficulty walking, speaking, with sensations, or unconsciousness.

Retinoblastoma (Rb) is a rare form of cancer that rapidly develops from the immature cells of a retina, the light-detecting tissue of the eye. It is the most common primary malignant intraocular cancer in children, and it is almost exclusively found in young children.

An ependymoma is a tumor that arises from the ependyma, a tissue of the central nervous system. Usually, in pediatric cases the location is intracranial, while in adults it is spinal. The common location of intracranial ependymomas is the fourth ventricle. Rarely, ependymomas can occur in the pelvic cavity.
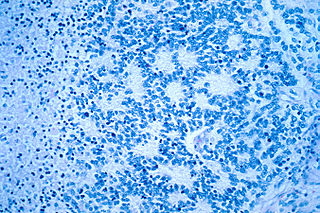
Neuroblastoma (NB) is a type of cancer that forms in certain types of nerve tissue. It most frequently starts from one of the adrenal glands but can also develop in the head, neck, chest, abdomen, or spine. Symptoms may include bone pain, a lump in the abdomen, neck, or chest, or a painless bluish lump under the skin.
A blastoma is a type of cancer, more common in children, that is caused by malignancies in precursor cells, often called blasts. Examples are nephroblastoma, medulloblastoma, and retinoblastoma. The suffix -blastoma is used to imply a tumor of primitive, incompletely differentiated cells, e.g., chondroblastoma is composed of cells resembling the precursor of chondrocytes.

Medulloblastoma is a common type of primary brain cancer in children. It originates in the part of the brain that is towards the back and the bottom, on the floor of the skull, in the cerebellum, or posterior fossa.

Primitive neuroectodermal tumor is a malignant (cancerous) neural crest tumor. It is a rare tumor, usually occurring in children and young adults under 25 years of age. The overall 5 year survival rate is about 53%.

An atypical teratoid rhabdoid tumor (AT/RT) is a rare tumor usually diagnosed in childhood. Although usually a brain tumor, AT/RT can occur anywhere in the central nervous system (CNS), including the spinal cord. About 60% will be in the posterior cranial fossa. One review estimated 52% in the posterior fossa, 39% are supratentorial primitive neuroectodermal tumors (sPNET), 5% are in the pineal, 2% are spinal, and 2% are multifocal.
The Ewing family of tumors (EFTs) is a group of small cell sarcomas including Ewing sarcoma of the bone, extra osseous Ewing tumors, and primitive neuroectodermal tumors. They are rare cancers, usually diagnosed in peoples' twenties. The sarcoma of bone is the most common of the variants. All forms are predisposed to metastasis and have had historically high rates of mortality. The family of tumors shares a common translocation mutation of the EWS gene on chromosome 22 to an ETS-type gene, most commonly the FLI1 gene. EFTs are highly malignant, with 5-year survival for patients with metastatic disease at 20%. The current standard of care includes resection, radiation, and chemotherapy.

Leptomeningeal cancer is a rare complication of cancer in which the disease spreads from the original tumor site to the meninges surrounding the brain and spinal cord. This leads to an inflammatory response, hence the alternative names neoplastic meningitis (NM), malignant meningitis, or carcinomatous meningitis. The term leptomeningeal describes the thin meninges, the arachnoid and the pia mater, between which the cerebrospinal fluid is located. The disorder was originally reported by Eberth in 1870.
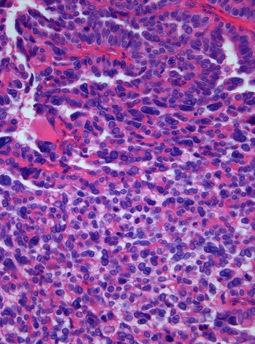
A choroid plexus carcinoma is a type of choroid plexus tumor that affects the choroid plexus of the brain. It is considered the worst of the three grades of chord plexus tumors, having a much poorer prognosis than choroid atypical plexus papilloma and choroid plexus papilloma. The disease creates lesions in the brain and increases cerebrospinal fluid volume, resulting in hydrocephalus.
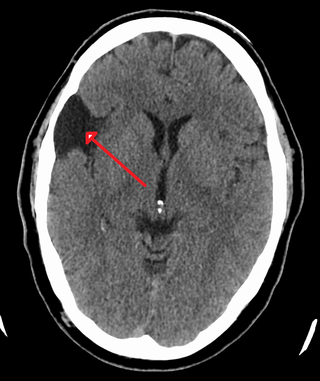
A central nervous system cyst is a type of cyst that presents and affects part of the central nervous system (CNS). They are usually benign and filled with either cerebrospinal fluid, blood, or tumor cells. CNS cysts are classified into two categories: cysts that originate from non-central nervous system tissue, migrate to, and form on a portion of the CNS, and cysts that originate within central nervous system tissue itself. Within these two categories, there are many types of CNS cysts that have been identified from previous studies.

Pineoblastoma is a malignant tumor of the pineal gland. A pineoblastoma is a supratentorial midline primitive neuroectodermal tumor. Pineoblastoma can present at any age, but is most common in young children. They account for 0.001% of all primary CNS neoplasms.

Medulloepithelioma is a rare, primitive, fast-growing brain tumour thought to stem from cells of the embryonic medullary cavity. Tumours originating in the ciliary body of the eye are referred to as embryonal medulloepitheliomas, or diktyomas.
Neuro-oncology is the study of brain and spinal cord neoplasms, many of which are very dangerous and life-threatening. Among the malignant brain cancers, gliomas of the brainstem and pons, glioblastoma multiforme, and high-grade astrocytoma/oligodendroglioma are among the worst. In these cases, untreated survival usually amounts to only a few months, and survival with current radiation and chemotherapy treatments may extend that time from around a year to a year and a half, possibly two or more, depending on the patient's condition, immune function, treatments used, and the specific type of malignant brain neoplasm. Surgery may in some cases be curative, but, as a general rule, malignant brain cancers tend to regenerate and emerge from remission easily, especially highly malignant cases. In such cases, the goal is to excise as much of the mass and as much of the tumor margin as possible without endangering vital functions or other important cognitive abilities. The Journal of Neuro-Oncology is the longest continuously published journal in the field and serves as a leading reference to those practicing in the area of neuro-oncology.
Embryonal rhabdomyosarcoma (EMRS) is a rare histological form of cancer in the connective tissue wherein the mesenchymally-derived malignant cells resemble the primitive developing skeletal muscle of the embryo. It is the most common soft tissue sarcoma occurring in children. Embryonal rhabdomyosarcoma is also known as PAX-fusion negative or fusion-negative rhabdomyosarcoma, as tumors of this subtype are unified by their lack of a PAX3-FOXO1 fusion oncogene. Fusion status refers to the presence or absence of a fusion gene, which is a gene formed from joining two different genes together through DNA rearrangements. These types of tumors are classified as embryonal rhabdomyosarcoma "because of their remarkable resemblance to developing embryonic and fetal skeletal muscle."
Bing–Neel syndrome (BNS) is an extremely rare neurologic complication of Waldenström macroglobulinemia (WM), which is a chronic lymphoproliferative disorder. There's no clear definition of BNS but what is known so far is that unlike WM, It involves the central nervous system (CNS), infiltrated by differentiated malignant B cells and by having hyperglobulinemia. This infiltration increases blood viscosity, which impairs blood circulation through small blood vessels of the brain and the eye. Some scientists proposed that a person diagnosed with BNS is typically classified into Group A and Group B depending on whether or not plasma cells are present within the brain parenchyma, leptomeninges, dura, and/or the cerebral spinal fluid (CSF). Symptoms are diverse and nonspecific, and they can vary depending on which aspect of the CNS is being affected. Symptoms can include a range of severity of nausea and seizures. Since the symptoms vary, there are multiple treatment options to treat the symptoms for this non-curable disease. Although there is no specific set of diagnosis for BNS, different combinations of diagnostic tools are used to narrow down and conclude the presence of BNS.

In histopathology, a palisade is a single layer of relatively long cells, arranged loosely perpendicular to a surface and parallel to each other. A rosette is a palisade in a halo or spoke-and-wheel arrangement, surrounding a central core or hub. A pseudorosette is a perivascular radial arrangement of neoplastic cells around a small blood vessel.
Embryonal tumor with multilayered rosettes (ETMR) is an embryonal central nervous system tumor. It is considered an embryonal tumor because it arises from cells partially differentiated or still undifferentiated from birth, usually neuroepithelial cells, stem cells destined to turn into glia or neurons. It can occur anywhere within the brain and can have multiple sites of origins, with a high probability of metastasis through cerebrospinal fluid (CSF). Metastases outside the central nervous system have been reported, but remain rare.



