
Thyroid neoplasm is a neoplasm or tumor of the thyroid. It can be a benign tumor such as thyroid adenoma, or it can be a malignant neoplasm, such as papillary, follicular, medullary or anaplastic thyroid cancer. Most patients are 25 to 65 years of age when first diagnosed; women are more affected than men. The estimated number of new cases of thyroid cancer in the United States in 2023 is 43,720 compared to only 2,120 deaths. Of all thyroid nodules discovered, only about 5 percent are cancerous, and under 3 percent of those result in fatalities.

Multiple endocrine neoplasia is a condition which encompasses several distinct syndromes featuring tumors of endocrine glands, each with its own characteristic pattern. In some cases, the tumors are malignant, in others, benign. Benign or malignant tumors of nonendocrine tissues occur as components of some of these tumor syndromes.
Hypercalcemia, also spelled hypercalcaemia, is a high calcium (Ca2+) level in the blood serum. The normal range is 2.1–2.6 mmol/L (8.8–10.7 mg/dL, 4.3–5.2 mEq/L), with levels greater than 2.6 mmol/L defined as hypercalcemia. Those with a mild increase that has developed slowly typically have no symptoms. In those with greater levels or rapid onset, symptoms may include abdominal pain, bone pain, confusion, depression, weakness, kidney stones or an abnormal heart rhythm including cardiac arrest.

Hyperparathyroidism is an increase in parathyroid hormone (PTH) levels in the blood. This occurs from a disorder either within the parathyroid glands or as response to external stimuli. Symptoms of hyperparathyroidism are caused by inappropriately normal or elevated blood calcium excreted from the bones and flowing into the blood stream in response to increased production of parathyroid hormone. In healthy people, when blood calcium levels are high, parathyroid hormone levels should be low. With long-standing hyperparathyroidism, the most common symptom is kidney stones. Other symptoms may include bone pain, weakness, depression, confusion, and increased urination. Both primary and secondary may result in osteoporosis.

Endocrine surgery is a surgical sub-speciality focusing on surgery of the endocrine glands, including the thyroid gland, the parathyroid glands, the adrenal glands, glands of the endocrine pancreas, and some neuroendocrine glands.

A paraganglioma is a rare neuroendocrine neoplasm that may develop at various body sites. When the same type of tumor is found in the adrenal gland, they are referred to as a pheochromocytoma. They are rare tumors, with an overall estimated incidence of 1 in 300,000. There is no test that determines benign from malignant tumors; long-term follow-up is therefore recommended for all individuals with paraganglioma.
Primary hyperparathyroidism is a medical condition where the parathyroid gland produce excess amounts of parathyroid hormone (PTH). The symptoms of the condition relate to the resulting elevated serum calcium (hypercalcemia), which can cause digestive symptoms, kidney stones, psychiatric abnormalities, and bone disease.

Multiple endocrine neoplasia type 1 (MEN-1) is one of a group of disorders, the multiple endocrine neoplasias, that affect the endocrine system through development of neoplastic lesions in pituitary, parathyroid gland and pancreas. Individuals suffering from this disorder are prone to developing multiple endocrine and nonendocrine tumors. It was first described by Paul Wermer in 1954.

Osteitis fibrosa cystica is a skeletal disorder resulting in a loss of bone mass, a weakening of the bones as their calcified supporting structures are replaced with fibrous tissue, and the formation of cyst-like brown tumors in and around the bone. Osteitis fibrosis cystica (OFC), also known as osteitis fibrosa, osteodystrophia fibrosa, and von Recklinghausen's disease of bone, is caused by hyperparathyroidism, which is a surplus of parathyroid hormone from over-active parathyroid glands. This surplus stimulates the activity of osteoclasts, cells that break down bone, in a process known as osteoclastic bone resorption. The hyperparathyroidism can be triggered by a parathyroid adenoma, hereditary factors, parathyroid carcinoma, or renal osteodystrophy. Osteoclastic bone resorption releases minerals, including calcium, from the bone into the bloodstream, causing both elevated blood calcium levels, and the structural changes which weaken the bone. The symptoms of the disease are the consequences of both the general softening of the bones and the excess calcium in the blood, and include bone fractures, kidney stones, nausea, moth-eaten appearance in the bones, appetite loss, and weight loss.

Papillary thyroid cancer is the most common type of thyroid cancer, representing 75 percent to 85 percent of all thyroid cancer cases. It occurs more frequently in women and presents in the 20–55 year age group. It is also the predominant cancer type in children with thyroid cancer, and in patients with thyroid cancer who have had previous radiation to the head and neck. It is often well-differentiated, slow-growing, and localized, although it can metastasize.

Neuroendocrine tumors (NETs) are neoplasms that arise from cells of the endocrine (hormonal) and nervous systems. They most commonly occur in the intestine, where they are often called carcinoid tumors, but they are also found in the pancreas, lung, and the rest of the body.

Endocrine diseases are disorders of the endocrine system. The branch of medicine associated with endocrine disorders is known as endocrinology.

The RETproto-oncogene encodes a receptor tyrosine kinase for members of the glial cell line-derived neurotrophic factor (GDNF) family of extracellular signalling molecules. RET loss of function mutations are associated with the development of Hirschsprung's disease, while gain of function mutations are associated with the development of various types of human cancer, including medullary thyroid carcinoma, multiple endocrine neoplasias type 2A and 2B, pheochromocytoma and parathyroid hyperplasia.

Menin is a protein that in humans is encoded by the MEN1 gene. Menin is a putative tumor suppressor associated with multiple endocrine neoplasia type 1 and has autosomal dominant inheritance. Variations in the MEN1 gene can cause pituitary adenomas, hyperparathyroidism, pancreatic neuroendocrine tumors, gastrinoma, and adrenocortical cancers.

Follicular thyroid cancer accounts for 15% of thyroid cancer and occurs more commonly in women over 50 years of age. Thyroglobulin (Tg) can be used as a tumor marker for well-differentiated follicular thyroid cancer. Thyroid follicular cells are the thyroid cells responsible for the production and secretion of thyroid hormones.
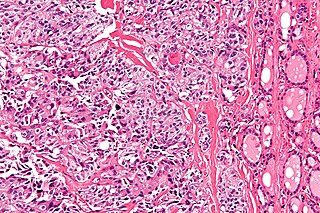
Medullary thyroid cancer is a form of thyroid carcinoma which originates from the parafollicular cells, which produce the hormone calcitonin. Medullary tumors are the third most common of all thyroid cancers and together make up about 3% of all thyroid cancer cases. MTC was first characterized in 1959.
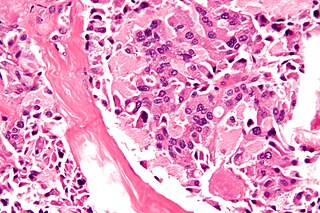
Multiple endocrine neoplasia type 2B is a genetic disease that causes multiple tumors on the mouth, eyes, and endocrine glands. It is the most severe type of multiple endocrine neoplasia, differentiated by the presence of benign oral and submucosal tumors in addition to endocrine malignancies. It was first described by Wagenmann in 1922, and was first recognized as a syndrome in 1965–1966 by E.D. Williams and D.J. Pollock. It is caused by the pathogenic variant p.Met918Thr in the RET gene. This variant can cause medullary thyroid cancer and Pheochromocytoma. Presentation can include a Marfanoid body, enlarged lips, and ganglionueuromas.

Parathyroid carcinoma is a rare cancer resulting in parathyroid adenoma to carcinoma progression. It forms in tissues of one or more of the parathyroid glands.
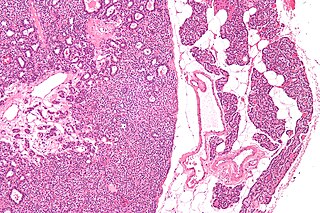
Many conditions are associated with disorders of the function of the parathyroid gland. Some disorders may be purely anatomical resulting in an enlarged gland which will raise concern. Such benign disorders, such as parathyroid cyst, are not discussed here. Parathyroid diseases can be divided into those causing hyperparathyroidism, and those causing hypoparathyroidism.
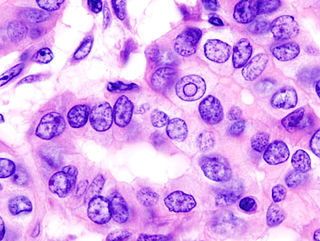
Thyroid cancer is cancer that develops from the tissues of the thyroid gland. It is a disease in which cells grow abnormally and have the potential to spread to other parts of the body. Symptoms can include swelling or a lump in the neck, difficulty swallowing or voice changes including hoarseness, or a feeling of something being in the throat due to mass effect from the tumor. However, most cases are asymptomatic. Cancer can also occur in the thyroid after spread from other locations, in which case it is not classified as thyroid cancer.


