Related Research Articles
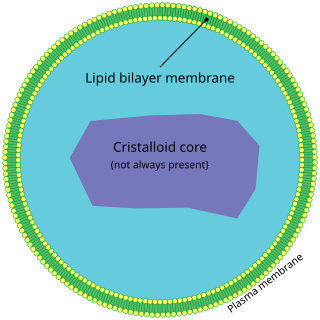
A peroxisome (IPA:[pɛɜˈɹɒksɪˌsoʊm]) is a membrane-bound organelle, a type of microbody, found in the cytoplasm of virtually all eukaryotic cells. Peroxisomes are oxidative organelles. Frequently, molecular oxygen serves as a co-substrate, from which hydrogen peroxide (H2O2) is then formed. Peroxisomes owe their name to hydrogen peroxide generating and scavenging activities. They perform key roles in lipid metabolism and the reduction of reactive oxygen species.
Enoyl-CoA-(∆) isomerase (EC 5.3.3.8, also known as dodecenoyl-CoA- isomerase, 3,2-trans-enoyl-CoA isomerase, ∆3 ,∆2 -enoyl-CoA isomerase, or acetylene-allene isomerase, is an enzyme that catalyzes the conversion of cis- or trans-double bonds of coenzyme A bound fatty acids at gamma-carbon to trans double bonds at beta-carbon as below:

Zellweger syndrome is a rare congenital disorder characterized by the reduction or absence of functional peroxisomes in the cells of an individual. It is one of a family of disorders called Zellweger spectrum disorders which are leukodystrophies. Zellweger syndrome is named after Hans Zellweger (1909–1990), a Swiss-American pediatrician, a professor of pediatrics and genetics at the University of Iowa who researched this disorder.
In biochemistry and metabolism, beta oxidation (also β-oxidation) is the catabolic process by which fatty acid molecules are broken down in the cytosol in prokaryotes and in the mitochondria in eukaryotes to generate acetyl-CoA. Acetyl-CoA enters the citric acid cycle, generating NADH and FADH2, which are electron carriers used in the electron transport chain. It is named as such because the beta carbon of the fatty acid chain undergoes oxidation and is converted to a carbonyl group to start the cycle all over again. Beta-oxidation is primarily facilitated by the mitochondrial trifunctional protein, an enzyme complex associated with the inner mitochondrial membrane, although very long chain fatty acids are oxidized in peroxisomes.

Mitochondrial trifunctional protein deficiency is an autosomal recessive fatty acid oxidation disorder that prevents the body from converting certain fats to energy, particularly during periods without food. People with this disorder have inadequate levels of an enzyme that breaks down a certain group of fats called long-chain fatty acids.
Refsum disease is an autosomal recessive neurological disease that results in the over-accumulation of phytanic acid in cells and tissues. It is one of several disorders named after Norwegian neurologist Sigvald Bernhard Refsum (1907–1991). Refsum disease typically is adolescent onset and is diagnosed by above average levels of phytanic acid. Humans obtain the necessary phytanic acid primarily through diet. It is still unclear what function phytanic acid plays physiologically in humans, but has been found to regulate fatty acid metabolism in the liver of mice.

Acyl-CoA dehydrogenase, C-2 to C-3 short chain is an enzyme that in humans is encoded by the ACADS gene. This gene encodes a tetrameric mitochondrial flavoprotein, which is a member of the acyl-CoA dehydrogenase family. This enzyme catalyzes the initial step of the mitochondrial fatty acid beta-oxidation pathway. The ACADS gene is associated with short-chain acyl-coenzyme A dehydrogenase deficiency.
Peroxisomal disorders represent a class of medical conditions caused by defects in peroxisome functions. This may be due to defects in single enzymes important for peroxisome function or in peroxins, proteins encoded by PEX genes that are critical for normal peroxisome assembly and biogenesis.

Malonyl-CoA decarboxylase, is found in bacteria and humans and has important roles in regulating fatty acid metabolism and food intake, and it is an attractive target for drug discovery. It is an enzyme associated with Malonyl-CoA decarboxylase deficiency. In humans, it is encoded by the MLYCD gene.

Trifunctional enzyme subunit alpha, mitochondrial also known as hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase/enoyl-CoA hydratase, alpha subunit is a protein that in humans is encoded by the HADHA gene. Mutations in HADHA have been associated with trifunctional protein deficiency or long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency.

2,4 Dienoyl-CoA reductase also known as DECR1 is an enzyme which in humans is encoded by the DECR1 gene which resides on chromosome 8. This enzyme catalyzes the following reactions

Thiolases, also known as acetyl-coenzyme A acetyltransferases (ACAT), are enzymes which convert two units of acetyl-CoA to acetoacetyl CoA in the mevalonate pathway.

ACADSB is a human gene that encodes short/branched chain specific acyl-CoA dehydrogenase (SBCAD), an enzyme in the acyl CoA dehydrogenase family.
Infantile Refsum disease (IRD) is a rare autosomal recessive congenital peroxisomal biogenesis disorder within the Zellweger spectrum. These are disorders of the peroxisomes that are clinically similar to Zellweger syndrome and associated with mutations in the PEX family of genes. IRD is associated with deficient phytanic acid catabolism, as is adult Refsum disease, but they are different disorders that should not be confused.
In enzymology, a phytanoyl-CoA dioxygenase (EC 1.14.11.18) is an enzyme that catalyzes the chemical reaction

α-Methylacyl-CoA racemase is an enzyme that in humans is encoded by the AMACR gene. AMACR catalyzes the following chemical reaction:

D-bifunctional protein (DBP), also known as peroxisomal multifunctional enzyme type 2 (MFP-2), as well as 17β-hydroxysteroid dehydrogenase type IV is a protein that in humans is encoded by the HSD17B4 gene. It's an alcohol oxidoreductase, specifically 17β-Hydroxysteroid dehydrogenase. It is involved in fatty acid β-oxidation and steroid metabolism.

ATP-binding cassette sub-family D member 3 is a protein that in humans is encoded by the ABCD3 gene.

Hydroxyacyl-Coenzyme A dehydrogenase (HADH) is an enzyme which in humans is encoded by the HADH gene.
EHHADH is a human gene that encodes for a bifunctional enzyme and is one of the four enzymes of the peroxisomal beta-oxidation pathway. Mutations of the gene are a cause of peroxisomal disorders such as Zellweger syndrome.
References
- 1 2 3 4 5 6 Möller G, van Grunsven EG, Wanders RJ, Adamski J (January 2001). "Molecular basis of D-bifunctional protein deficiency". Mol. Cell. Endocrinol. 171 (1–2): 61–70. doi:10.1016/s0303-7207(00)00388-9. PMID 11165012. S2CID 29712091.
- ↑ Itoh M, Suzuki Y, Akaboshi S, Zhang Z, Miyabara S, Takashima S (March 2000). "Developmental and pathological expression of peroxisomal enzymes: their relationship of D-bifunctional protein deficiency and Zellweger syndrome". Brain Res. 858 (1): 40–7. doi:10.1016/S0006-8993(99)02423-3. PMID 10700594. S2CID 11224543.
- ↑ Buoni S, Zannolli R, Waterham H, Wanders R, Fois A (January 2007). "D-bifunctional protein deficiency associated with drug resistant infantile spasms". Brain Dev. 29 (1): 51–4. doi:10.1016/j.braindev.2006.06.004. PMID 16919904. S2CID 617635.
- 1 2 3 4 5 6 Ferdinandusse S, Ylianttila MS, Gloerich J, Koski MK, Oostheim W, Waterham HR, Hiltunen JK, Wanders RJ, Glumoff T (January 2006). "Mutational spectrum of D-bifunctional protein deficiency and structure-based genotype-phenotype analysis". Am. J. Hum. Genet. 78 (1): 112–24. doi:10.1086/498880. PMC 1380208 . PMID 16385454.
- ↑ van Grunsven EG, Mooijer PA, Aubourg P, Wanders RJ (August 1999). "Enoyl-CoA hydratase deficiency: identification of a new type of D-bifunctional protein deficiency". Hum. Mol. Genet. 8 (8): 1509–16. doi: 10.1093/hmg/8.8.1509 . PMID 10400999.