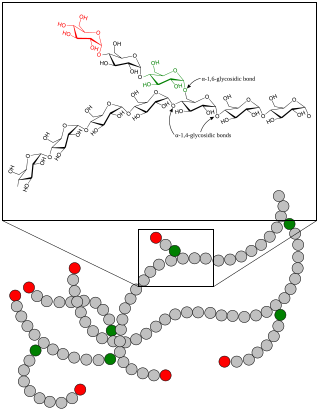
A glycogen storage disease is a metabolic disorder caused by a deficiency of an enzyme or transport protein affecting glycogen synthesis, glycogen breakdown, or glucose breakdown, typically in muscles and/or liver cells.

Gaucher's disease or Gaucher disease (GD) is a genetic disorder in which glucocerebroside accumulates in cells and certain organs. The disorder is characterized by bruising, fatigue, anemia, low blood platelet count and enlargement of the liver and spleen, and is caused by a hereditary deficiency of the enzyme glucocerebrosidase, which acts on glucocerebroside. When the enzyme is defective, glucocerebroside accumulates, particularly in white blood cells and especially in macrophages. Glucocerebroside can collect in the spleen, liver, kidneys, lungs, brain, and bone marrow.

Glycogen storage disease type II(GSD-II), also called Pompe disease, and formerly known as GSD-IIa or Limb–girdle muscular dystrophy2V, is an autosomal recessive metabolic disorder which damages muscle and nerve cells throughout the body. It is caused by an accumulation of glycogen in the lysosome due to deficiency of the lysosomal acid alpha-glucosidase enzyme (GAA). The inability to breakdown glycogen within the lysosomes of cells leads to progressive muscle weakness throughout the body and affects various body tissues, particularly in the heart, skeletal muscles, liver and the nervous system.

Phosphofructokinase deficiency is a rare muscular metabolic disorder, with an autosomal recessive inheritance pattern.
Inborn errors of metabolism form a large class of genetic diseases involving congenital disorders of enzyme activities. The majority are due to defects of single genes that code for enzymes that facilitate conversion of various substances (substrates) into others (products). In most of the disorders, problems arise due to accumulation of substances which are toxic or interfere with normal function, or due to the effects of reduced ability to synthesize essential compounds. Inborn errors of metabolism are often referred to as congenital metabolic diseases or inherited metabolic disorders. Another term used to describe these disorders is "enzymopathies". This term was created following the study of biodynamic enzymology, a science based on the study of the enzymes and their products. Finally, inborn errors of metabolism were studied for the first time by British physician Archibald Garrod (1857–1936), in 1908. He is known for work that prefigured the "one gene–one enzyme" hypothesis, based on his studies on the nature and inheritance of alkaptonuria. His seminal text, Inborn Errors of Metabolism, was published in 1923.

Glycogen phosphorylase is one of the phosphorylase enzymes. Glycogen phosphorylase catalyzes the rate-limiting step in glycogenolysis in animals by releasing glucose-1-phosphate from the terminal alpha-1,4-glycosidic bond. Glycogen phosphorylase is also studied as a model protein regulated by both reversible phosphorylation and allosteric effects.

Glycogen storage disease type I is an inherited disease that prevents the liver from properly breaking down stored glycogen, which is necessary to maintain adequate blood sugar levels. GSD I is divided into two main types, GSD Ia and GSD Ib, which differ in cause, presentation, and treatment. There are also possibly rarer subtypes, the translocases for inorganic phosphate or glucose ; however, a recent study suggests that the biochemical assays used to differentiate GSD Ic and GSD Id from GSD Ib are not reliable, and are therefore GSD Ib.

The glycogen debranching enzyme, in humans, is the protein encoded by the gene AGL. This enzyme is essential for the breakdown of glycogen, which serves as a store of glucose in the body. It has separate glucosyltransferase and glucosidase activities.
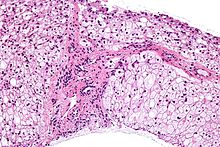
Glycogen storage disease type IV (GSD IV), or Andersen's Disease, is a form of glycogen storage disease, which is caused by an inborn error of metabolism. It is the result of a mutation in the GBE1 gene, which causes a defect in the glycogen branching enzyme. Therefore, glycogen is not made properly and abnormal glycogen molecules accumulate in cells; most severely in cardiac and muscle cells. The severity of this disease varies on the amount of enzyme produced. GSD IV is autosomal recessive, which means each parent has a mutant copy of the gene, but show no symptoms of the disease. Having an autosomal recessive inheritance pattern, males and females are equally likely to be affected by Andersen's disease. Classic Andersen's disease typically becomes apparent during the first few months after the patient is born. Approximately 1 in 20,000 to 25,000 newborns have a glycogen storage disease. Andersen's disease affects 1 in 800,000 individuals worldwide, with 3% of all GSDs being type IV. The disease was described and studied first by Dorothy Hansine Andersen.

Glycogen storage disease type 0 is a disease characterized by a deficiency in the glycogen synthase enzyme (GSY). Although glycogen synthase deficiency does not result in storage of extra glycogen in the liver, it is often classified as a glycogen storage disease because it is another defect of glycogen storage and can cause similar problems. There are two isoforms (types) of glycogen synthase enzyme; GSY1 in muscle and GSY2 in liver, each with a corresponding form of the disease. Mutations in the liver isoform (GSY2), causes fasting hypoglycemia, high blood ketones, increased free fatty acids and low levels of alanine and lactate. Conversely, feeding in these patients results in hyperglycemia and hyperlactatemia.

Boron, Walter F.; Boulpaep, Emile L., eds. (2017). Medical Physiology (3rd ed.). Philadelphia, PA: Elsevier. ISBN 978-1-4557-4377-3.

1,4-alpha-glucan-branching enzyme, also known as brancher enzyme or glycogen-branching enzyme is an enzyme that in humans is encoded by the GBE1 gene.

Acid alpha-glucosidase, also called acid maltase, is an enzyme that helps to break down glycogen in the lysosome. It is functionally similar to glycogen debranching enzyme, but is on a different chromosome, processed differently by the cell and is located in the lysosome rather than the cytosol. In humans, it is encoded by the GAA gene. Errors in this gene cause glycogen storage disease type II.

Myophosphorylase or glycogen phosphorylase, muscle associated (PYGM) is the muscle isoform of the enzyme glycogen phosphorylase and is encoded by the PYGM gene. This enzyme helps break down glycogen into glucose-1-phosphate, so it can be used within the muscle cell. Mutations in this gene are associated with McArdle disease, a glycogen storage disease of muscle.

Phosphorylase b kinase gamma catalytic chain, testis/liver isoform is an enzyme that in humans is encoded by the PHKG2 gene.
Glycogen storage disease type IX is a hereditary deficiency of glycogen phosphorylase kinase B that affects the liver and skeletal muscle tissue. It is inherited in an X-linked or autosomal recessive manner.

Inborn errors of carbohydrate metabolism are inborn error of metabolism that affect the catabolism and anabolism of carbohydrates.
Danon disease is a metabolic disorder. Danon disease is an X-linked lysosomal and glycogen storage disorder associated with hypertrophic cardiomyopathy, skeletal muscle weakness, and intellectual disability. It is inherited in an X-linked dominant pattern.

Metabolic myopathies are myopathies that result from defects in biochemical metabolism that primarily affect muscle. They are generally genetic defects that interfere with the ability to create energy, causing a low ATP reservoir within the muscle cell.
Barbara Illingworth Brown was an American biochemist. She worked primarily at Washington University in St. Louis.