| Pentosuria | |
|---|---|
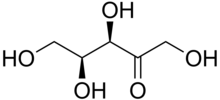 | |
| Xylulose | |
| Specialty | Endocrinology |
Pentosuria is a condition where the sugar L-xylulose, a pentose, presents in the urine in unusually high concentrations. It was characterized as an inborn error of carbohydrate metabolism in 1908. [1] It is associated with a deficiency of L-xylulose reductase, necessary for xylitol metabolism. [2] [3] L-Xylulose is a reducing sugar, so it may give false diagnosis of diabetes, as it is found in high concentrations in urine. However glucose metabolism is normal in people with pentosuria, and they are not diabetic. [4] Patients of pentosuria have a low concentration of the sugar d-xyloketose. [5] Using phenyl pentosazone crystals, phloroglucin reaction, and absorption spectrum, pentose can be traced back as the reducing substance in urine, with those that have pentosuria. [6]
Research has shown that pentosuria appears in 3 forms. The most widely studied is essential pentosuria, where a couple of grams of L-xylulose are released into a person's system daily. [7] L-xylulose reductase, contained in red blood cells, is composed of both a major and minor isozyme. [8] For those diagnosed with essential pentosuria, the major isozyme appears to be the same as the minor one. [8] Alimentary pentosuria can be acquired through fruits high in pentose. [7] Finally, drug-induced pentosuria can be developed by those exposed to morphine, fevers, allergies, and some hormones. [7]
Those diagnosed with Pentosuria are predominantly of Jewish heritage. [2] It is a harmless defect, and no cure is needed. [9]