Galactose, sometimes abbreviated Gal, is a monosaccharide sugar that is about as sweet as glucose, and about 65% as sweet as sucrose. It is an aldohexose and a C-4 epimer of glucose. A galactose molecule linked with a glucose molecule forms a lactose molecule.

Galactosemia is a rare genetic metabolic disorder that affects an individual's ability to metabolize the sugar galactose properly. Galactosemia follows an autosomal recessive mode of inheritance that confers a deficiency in an enzyme responsible for adequate galactose degradation.

In biochemistry, a transferase is any one of a class of enzymes that catalyse the transfer of specific functional groups from one molecule to another. They are involved in hundreds of different biochemical pathways throughout biology, and are integral to some of life's most important processes.
Galactokinase is an enzyme (phosphotransferase) that facilitates the phosphorylation of α-D-galactose to galactose 1-phosphate at the expense of one molecule of ATP. Galactokinase catalyzes the second step of the Leloir pathway, a metabolic pathway found in most organisms for the catabolism of α-D-galactose to glucose 1-phosphate. First isolated from mammalian liver, galactokinase has been studied extensively in yeast, archaea, plants, and humans.

Galactose-1-phosphate uridyltransferase is an enzyme responsible for converting ingested galactose to glucose.

Galactokinase deficiency is an autosomal recessive metabolic disorder marked by an accumulation of galactose and galactitol secondary to the decreased conversion of galactose to galactose-1-phosphate by galactokinase. The disorder is caused by mutations in the GALK1 gene, located on chromosome 17q24. Galactokinase catalyzes the first step of galactose phosphorylation in the Leloir pathway of intermediate metabolism. Galactokinase deficiency is one of the three inborn errors of metabolism that lead to hypergalactosemia. The disorder is inherited as an autosomal recessive trait. Unlike classic galactosemia, which is caused by a deficiency of galactose-1-phosphate uridyltransferase, galactokinase deficiency does not present with severe manifestations in early infancy. Its major clinical symptom is the development of cataracts during the first weeks or months of life, as a result of the accumulation, in the lens, of galactitol, a product of an alternative route of galactose utilization. The development of early cataracts in homozygous affected infants is fully preventable through early diagnosis and treatment with a galactose-restricted diet. Some studies have suggested that, depending on milk consumption later in life, heterozygous carriers of galactokinase deficiency may be prone to presenile cataracts at 20–50 years of age.
UTP—glucose-1-phosphate uridylyltransferase also known as glucose-1-phosphate uridylyltransferase is an enzyme involved in carbohydrate metabolism. It synthesizes UDP-glucose from glucose-1-phosphate and UTP; i.e.,

Galactose epimerase deficiency, also known as GALE deficiency, Galactosemia III and UDP-galactose-4-epimerase deficiency, is a rare, autosomal recessive form of galactosemia associated with a deficiency of the enzyme galactose epimerase.
Uridine diphosphate galactose (UDP-galactose) is an intermediate in the production of polysaccharides. It is important in nucleotide sugars metabolism, and is the substrate for the transferase B4GALT5.

D-Galactose-1-phosphate is an intermediate in the intraconversion of glucose and uridine diphosphate galactose. It is formed from galactose by galactokinase.The improper metabolism of galactose-1-phosphate is a characteristic of galactosemia. The Leloir pathway is responsible for such metabolism of galactose and its intermediate, galactose-1-phosphate. Deficiency of enzymes found in this pathway can result in galactosemia; therefore, diagnosis of this genetic disorder occasionally involves measuring the concentration of these enzymes. One of such enzymes is galactose-1-phosphate uridylyltransferase (GALT). The enzyme catalyzes the transfer of a UDP-activator group from UDP-glucose to galactose-1-phosphate. Although the cause of enzyme deficiency in the Leloir pathway is still disputed amongst researchers, some studies suggest that protein misfolding of GALT, which may lead to an unfavorable conformational change that impacts its thermal stability and substrate-binding affinity, may play a role in the deficiency of GALT in Type 1 galactosemia. Increase in galactitol concentration can be seen in patients with galactosemia; putting patients at higher risk for presenile cataract.
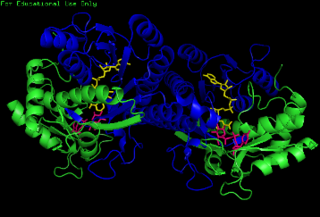
The enzyme UDP-glucose 4-epimerase, also known as UDP-galactose 4-epimerase or GALE, is a homodimeric epimerase found in bacterial, fungal, plant, and mammalian cells. This enzyme performs the final step in the Leloir pathway of galactose metabolism, catalyzing the reversible conversion of UDP-galactose to UDP-glucose. GALE tightly binds nicotinamide adenine dinucleotide (NAD+), a co-factor required for catalytic activity.
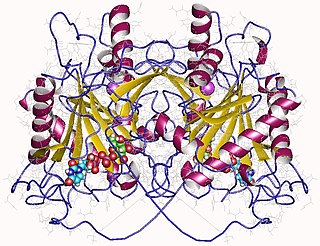
In enzymology, an UDP-glucose—hexose-1-phosphate uridylyltransferase is an enzyme that catalyzes the chemical reaction
The gal operon is a prokaryotic operon, which encodes enzymes necessary for galactose metabolism. Repression of gene expression for this operon works via binding of repressor molecules to two operators. These repressors dimerize, creating a loop in the DNA. The loop as well as hindrance from the external operator prevent RNA polymerase from binding to the promoter, and thus prevent transcription. Additionally, since the metabolism of galactose in the cell is involved in both anabolic and catabolic pathways, a novel regulatory system using two promoters for differential repression has been identified and characterized within the context of the gal operon.
A galactosemic cataract is cataract which is associated with the consequences of galactosemia.

Inborn errors of carbohydrate metabolism are inborn error of metabolism that affect the catabolism and anabolism of carbohydrates.
Galactitol (dulcitol) is a sugar alcohol, the reduction product of galactose. It has a slightly sweet taste. In people with galactokinase deficiency, a form of galactosemia, excess dulcitol forms in the lens of the eye leading to cataracts.
Galactolysis refers to the catabolism of galactose.
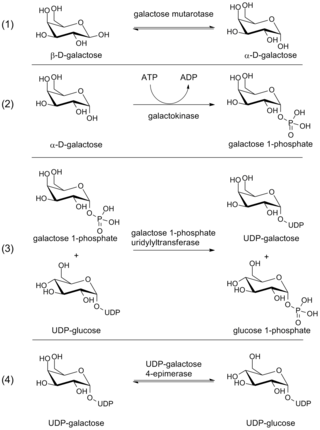
The Leloir pathway is a metabolic pathway for the catabolism of D-galactose. It is named after Luis Federico Leloir, who first described it.

Duarte galactosemia is an inherited condition associated with diminished ability to metabolize galactose due to a partial deficiency of the enzyme galactose-1-phosphate uridylyltransferase. DG differs from classic galactosemia in that patients with Duarte galactosemia have partial GALT deficiency whereas patients with classic galactosemia have complete, or almost complete, GALT deficiency. Duarte galactosemia (DG) is much more common than classic galactosemia, and is estimated to affect close to one in 4,000 infants born in the United States. Historically, most healthcare professionals have considered DG to be clinically mild based on pilot studies and anecdotal experience, and in 2019 a large study confirmed that children with DG are not at increased risk for developmental problems relative to children who do not have DG. Due to regional variations in newborn screening (NBS) protocols, some infants with DG are identified by NBS but others are not.



