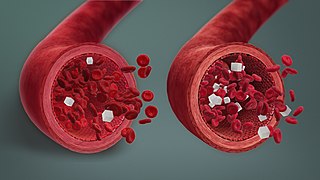
Hypoglycemia, also spelled hypoglycaemia or hypoglycæmia, sometimes called low blood sugar, is a fall in blood sugar to levels below normal, typically below 70 mg/dL (3.9 mmol/L). Whipple's triad is used to properly identify hypoglycemic episodes. It is defined as blood glucose below 70 mg/dL (3.9 mmol/L), symptoms associated with hypoglycemia, and resolution of symptoms when blood sugar returns to normal. Hypoglycemia may result in headache, tiredness, clumsiness, trouble talking, confusion, fast heart rate, sweating, shakiness, nervousness, hunger, loss of consciousness, seizures, or death. Symptoms typically come on quickly.

A glycogen storage disease is a metabolic disorder caused by a deficiency of an enzyme or transport protein affecting glycogen synthesis, glycogen breakdown, or glucose breakdown, typically in muscles and/or liver cells.
Gluconeogenesis (GNG) is a metabolic pathway that results in the biosynthesis of glucose from certain non-carbohydrate carbon substrates. It is a ubiquitous process, present in plants, animals, fungi, bacteria, and other microorganisms. In vertebrates, gluconeogenesis occurs mainly in the liver and, to a lesser extent, in the cortex of the kidneys. It is one of two primary mechanisms – the other being degradation of glycogen (glycogenolysis) – used by humans and many other animals to maintain blood sugar levels, avoiding low levels (hypoglycemia). In ruminants, because dietary carbohydrates tend to be metabolized by rumen organisms, gluconeogenesis occurs regardless of fasting, low-carbohydrate diets, exercise, etc. In many other animals, the process occurs during periods of fasting, starvation, low-carbohydrate diets, or intense exercise.

Glucagon is a peptide hormone, produced by alpha cells of the pancreas. It raises the concentration of glucose and fatty acids in the bloodstream and is considered to be the main catabolic hormone of the body. It is also used as a medication to treat a number of health conditions. Its effect is opposite to that of insulin, which lowers extracellular glucose. It is produced from proglucagon, encoded by the GCG gene.

The blood sugar level, blood sugar concentration, blood glucose level, or glycemia is the measure of glucose concentrated in the blood. The body tightly regulates blood glucose levels as a part of metabolic homeostasis.

In fructose bisphosphatase deficiency, there is not enough fructose bisphosphatase for gluconeogenesis to occur correctly. Glycolysis will still work, as it does not use this enzyme.
Carbohydrate metabolism is the whole of the biochemical processes responsible for the metabolic formation, breakdown, and interconversion of carbohydrates in living organisms.
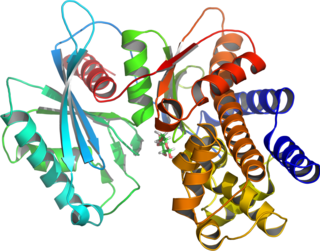
Glucokinase is an enzyme that facilitates phosphorylation of glucose to glucose-6-phosphate. Glucokinase occurs in cells in the liver and pancreas of humans and most other vertebrates. In each of these organs it plays an important role in the regulation of carbohydrate metabolism by acting as a glucose sensor, triggering shifts in metabolism or cell function in response to rising or falling levels of glucose, such as occur after a meal or when fasting. Mutations of the gene for this enzyme can cause unusual forms of diabetes or hypoglycemia.

Phosphofructokinase deficiency is a rare muscular metabolic disorder, with an autosomal recessive inheritance pattern. It is characterized as a deficiency in the Phosphofructokinase (PFK) enzyme throughout the body, including the skeletal muscles and red blood cells. Phosphofrucotkinase is an enzyme involved in the glycolytic process. The lack of PFK blocks the completion of the glycolytic pathway. Therefore, all products past the block would be deficient, including Adenosine triphosphate (ATP).
Ketotic hypoglycemia refers to any circumstance in which low blood glucose is accompanied by ketosis, the presence of ketone bodies in the blood or urine. This state can be either physiologic or pathologic; physiologic ketotic hypoglycemia is a common cause of hypoglycemia in children, often in response to stressors such as infection or fasting. Pathologic ketotic hypoglycemia is typically caused by metabolic defects, such as glycogen storage disorders.

Glycogen phosphorylase is one of the phosphorylase enzymes. Glycogen phosphorylase catalyzes the rate-limiting step in glycogenolysis in animals by releasing glucose-1-phosphate from the terminal alpha-1,4-glycosidic bond. Glycogen phosphorylase is also studied as a model protein regulated by both reversible phosphorylation and allosteric effects.

Glycogen storage disease type I is an inherited disease that prevents the liver from properly breaking down stored glycogen, which is necessary to maintain adequate blood sugar levels. GSD I is divided into two main types, GSD Ia and GSD Ib, which differ in cause, presentation, and treatment. There are also possibly rarer subtypes, the translocases for inorganic phosphate or glucose ; however, a recent study suggests that the biochemical assays used to differentiate GSD Ic and GSD Id from GSD Ib are not reliable, and are therefore GSD Ib.

Diabetic hypoglycemia is a low blood glucose level occurring in a person with diabetes mellitus. It is one of the most common types of hypoglycemia seen in emergency departments and hospitals. According to the National Electronic Injury Surveillance System-All Injury Program (NEISS-AIP), and based on a sample examined between 2004 and 2005, an estimated 55,819 cases involved insulin, and severe hypoglycemia is likely the single most common event.

Reactive hypoglycemia, postprandial hypoglycemia, or sugar crash is a term describing recurrent episodes of symptomatic hypoglycemia occurring within four hours after a high carbohydrate meal in people with and without diabetes. The term is not necessarily a diagnosis since it requires an evaluation to determine the cause of the hypoglycemia.
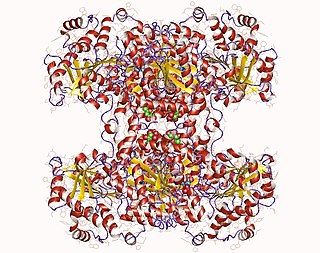
Glycogen synthase is a key enzyme in glycogenesis, the conversion of glucose into glycogen. It is a glycosyltransferase that catalyses the reaction of UDP-glucose and n to yield UDP and n+1.

The glycogen debranching enzyme, in humans, is the protein encoded by the gene AGL. This enzyme is essential for the breakdown of glycogen, which serves as a store of glucose in the body. It has separate glucosyltransferase and glucosidase activities.
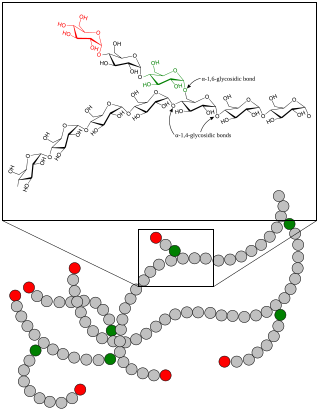
Glycogen storage disease type VI is a type of glycogen storage disease caused by a deficiency in liver glycogen phosphorylase or other components of the associated phosphorylase cascade system. It is also known as "Hers' disease", after Henri G. Hers, who characterized it in 1959. The scope of GSD VI now also includes glycogen storage disease type VIII, IX and X.

Inborn errors of carbohydrate metabolism are inborn error of metabolism that affect the catabolism and anabolism of carbohydrates.
Glycogen phosphorylase, liver form (PYGL), also known as human liver glycogen phosphorylase (HLGP), is an enzyme that in humans is encoded by the PYGL gene on chromosome 14. This gene encodes a homodimeric protein that catalyses the cleavage of alpha-1,4-glucosidic bonds to release glucose-1-phosphate from liver glycogen stores. This protein switches from inactive phosphorylase B to active phosphorylase A by phosphorylation of serine residue 14. Activity of this enzyme is further regulated by multiple allosteric effectors and hormonal controls. Humans have three glycogen phosphorylase genes that encode distinct isozymes that are primarily expressed in liver, brain and muscle, respectively. The liver isozyme serves the glycemic demands of the body in general while the brain and muscle isozymes supply just those tissues. In glycogen storage disease type VI, also known as Hers disease, mutations in liver glycogen phosphorylase inhibit the conversion of glycogen to glucose and results in moderate hypoglycemia, mild ketosis, growth retardation and hepatomegaly. Alternative splicing results in multiple transcript variants encoding different isoforms [provided by RefSeq, Feb 2011].
