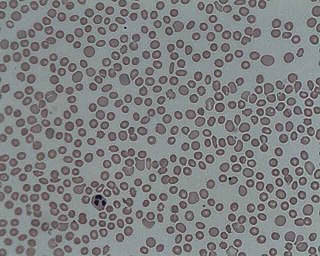
Thrombocytopenia is a condition characterized by abnormally low levels of platelets, also known as thrombocytes, in the blood. It is the most common coagulation disorder among intensive care patients and is seen in 20% of medical patients and a third of surgical patients.

Autoimmune polyendocrine syndromes (APSs), also called polyglandular autoimmune syndromes (PGASs) or polyendocrine autoimmune syndromes (PASs), are a heterogeneous group of rare diseases characterized by autoimmune activity against more than one endocrine organ, although non-endocrine organs can be affected. There are three types of APS, and there are a number of other diseases which involve endocrine autoimmunity.

Glanzmann's thrombasthenia is an abnormality of the platelets. It is an extremely rare coagulopathy, in which the platelets contain defective or low levels of glycoprotein IIb/IIIa (GpIIb/IIIa), which is a receptor for fibrinogen. As a result, no fibrinogen bridging of platelets to other platelets can occur, and the bleeding time is significantly prolonged.

Gardner's syndrome is a subtype of familial adenomatous polyposis (FAP). Gardner syndrome is an autosomal dominant form of polyposis characterized by the presence of multiple polyps in the colon together with tumors outside the colon. The extracolonic tumors may include osteomas of the skull, thyroid cancer, epidermoid cysts, fibromas, as well as the occurrence of desmoid tumors in approximately 15% of affected individuals.
Malouf syndrome is a congenital disorder that causes one or more of the following symptoms: mental retardation, ovarian dysgenesis, congestive cardiomyopathy, broad nasal base, blepharoptosis, and bone abnormalities, and occasionally marfanoid habitus.
Evans syndrome is an autoimmune disease in which an individual's immune system attacks their own red blood cells and platelets, the syndrome can include immune neutropenia. These immune cytopenias may occur simultaneously or sequentially.
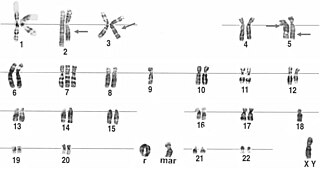
A ring chromosome is an aberrant chromosome whose ends have fused together to form a ring. Ring chromosomes were first discovered by Lilian Vaughan Morgan in 1926. A ring chromosome is denoted by the symbol r in human genetics and R in Drosophila genetics. Ring chromosomes may form in cells following genetic damage by mutagens like radiation, but they may also arise spontaneously during development.
Severe congenital neutropenia (SCN), also often known as Kostmann syndrome or disease, is a group of rare disorders that affect myelopoiesis, causing a congenital form of neutropenia, usually without other physical malformations. SCN manifests in infancy with life-threatening bacterial infections.
Heřmanský–Pudlák syndrome is an extremely rare autosomal recessive disorder which results in oculocutaneous albinism, bleeding problems due to a platelet abnormality, and storage of an abnormal fat-protein compound. It is considered to affect around 1 in 500,000 people worldwide, with a significantly higher occurrence in Puerto Ricans, with a prevalence of 1 in 1800. Many of the clinical research studies on the disease have been conducted in Puerto Rico.
Bernard–Soulier syndrome (BSS), is a rare autosomal recessive bleeding disorder that is caused by a deficiency of the glycoprotein Ib-IX-V complex (GPIb-IX-V), the receptor for von Willebrand factor. The incidence of BSS is estimated to be less than 1 case per million persons, based on cases reported from Europe, North America, and Japan. BSS is a giant platelet disorder, meaning that it is characterized by abnormally large platelets.

Griscelli syndrome is a rare autosomal recessive disorder characterized by albinism (hypopigmentation) with immunodeficiency, that usually causes death by early childhood. Researchers have developed three different classifications of the form of disorder, characterised by different signs and symptoms. Type 1 Griscelli Syndrome is assosciated with severe brain function issues along with distinctive discolouring of the hair and skin. Type 2 Griscelli Syndrome have immune system abnormalities in addition to hypopigmentation of skin and hair. Finally, Type 3 is seen as those only affected by hypopigmentation of the skin and hair. This type is not associated with immune deficiencies or neurological abnormalities.

GATA-binding factor 1 or GATA-1 is the founding member of the GATA family of transcription factors. This protein is widely expressed throughout vertebrate species. In humans and mice, it is encoded by the GATA1 and Gata1 genes, respectively. These genes are located on the X chromosome in both species.
Lucey–Driscoll syndrome is an autosomal recessive metabolic disorder affecting enzymes involved in bilirubin metabolism. It is one of several disorders classified as a transient familial neonatal unconjugated hyperbilirubinemia.

TNF receptor associated periodic syndrome (TRAPS) is a periodic fever syndrome associated with mutations in a receptor for the molecule tumor necrosis factor (TNF) that is inheritable in an autosomal dominant manner. Individuals with TRAPS have episodic symptoms such as recurrent high fevers, rash, abdominal pain, joint/muscle aches and puffy eyes.
Naegeli–Franceschetti–Jadassohn syndrome (NFJS), also known as chromatophore nevus of Naegeli and Naegeli syndrome, is a rare autosomal dominant form of ectodermal dysplasia, characterized by reticular skin pigmentation, diminished function of the sweat glands, the absence of teeth and hyperkeratosis of the palms and soles. One of the most striking features is the absence of fingerprint lines on the fingers.
Pseudo gray platelet syndrome was described by Cockbill, Burmester, and Heptinstall (1988) who reported a 25-year-old woman with a history of mild bruising and bleeding. Another case was described in Japan in 2002.
Platelet storage pool deficiency is a type of coagulopathy characterized by defects in the granules in platelets, particularly a lack of granular non-metabolic adenosine diphosphate. Individuals with adenosine diphosphate deficient storage pool disease present a prolonged bleeding time due to impaired aggregation response to fibrillar collagen.
Paris-Trousseau syndrome (PTS) is an inherited disorder characterized by mild hemorrhagic tendency associated with 11q chromosome deletion. It manifests as a granular defect within an individual's platelets. It is characterized by thrombocytes with defects in α-granule components which affects the cell's surface area and, consequently, its ability to spread when necessary.

Giant platelet disorders, also known as macrothrombocytopenia, are rare disorders featuring abnormally large platelets, thrombocytopenia and a tendency to bleeding. Giant platelets cannot stick adequately to an injured blood vessel walls, resulting in abnormal bleeding when injured. Giant platelet disorder occurs for inherited diseases like Bernard–Soulier syndrome, gray platelet syndrome and May–Hegglin anomaly.
