
A myelodysplastic syndrome (MDS) is one of a group of cancers in which immature blood cells in the bone marrow do not mature, and as a result, do not develop into healthy blood cells. Early on, no symptoms typically are seen. Later, symptoms may include fatigue, shortness of breath, bleeding disorders, anemia, or frequent infections. Some types may develop into acute myeloid leukemia.

Von Willebrand disease (VWD) is the most common hereditary blood-clotting disorder in humans. An acquired form can sometimes result from other medical conditions. It arises from a deficiency in the quality or quantity of von Willebrand factor (VWF), a multimeric protein that is required for platelet adhesion. It is known to affect several breeds of dogs as well as humans. The three forms of VWD are hereditary, acquired, and pseudo or platelet type. The three types of hereditary VWD are VWD type 1, VWD type 2, and VWD type 3. Type 2 contains various subtypes. Platelet type VWD is also an inherited condition.

Immune thrombocytopenic purpura (ITP), also known as idiopathic thrombocytopenic purpura or immune thrombocytopenia, is a type of thrombocytopenic purpura characterized by a low platelet count in the absence of other causes, and accompanied by a red-purple rash called purpura. It leads to an increased risk of bleeding. ITP manifests in two distinct clinical syndromes: an acute form observed in children, and chronic conditions observed in adults. The acute form often follows an infection and typically resolves within two months, while chronic immune thrombocytopenia persists for longer than six months and its specific cause is unknown.

Thrombotic thrombocytopenic purpura (TTP) is a blood disorder that results in blood clots forming in small blood vessels throughout the body. This results in a low platelet count, low red blood cells due to their breakdown, and often kidney, heart, and brain dysfunction. Symptoms may include large bruises, fever, weakness, shortness of breath, confusion, and headache. Repeated episodes may occur.
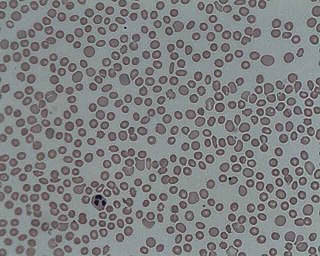
Thrombocytopenia is a condition characterized by abnormally low levels of platelets, also known as thrombocytes, in the blood. Low levels of platelets in turn may lead to prolonged or excessive bleeding. It is the most common coagulation disorder among intensive care patients and is seen in a fifth of medical patients and a third of surgical patients.

Hemolytic–uremic syndrome (HUS) is a group of blood disorders characterized by low red blood cells, acute kidney failure, and low platelets. Initial symptoms typically include bloody diarrhea, fever, vomiting, and weakness. Kidney problems and low platelets then occur as the diarrhea progresses. Children are more commonly affected, but most children recover without permanent damage to their health, although some children may have serious and sometimes life-threatening complications. Adults, especially the elderly, may present a more complicated presentation. Complications may include neurological problems and heart failure.

In medicine (hematology), bleeding diathesis is an unusual susceptibility to bleed (hemorrhage) mostly due to hypocoagulability, in turn caused by a coagulopathy. Therefore, this may result in the reduction of platelets being produced and leads to excessive bleeding. Several types of coagulopathy are distinguished, ranging from mild to lethal. Coagulopathy can be caused by thinning of the skin, such that the skin is weakened and is bruised easily and frequently without any trauma or injury to the body. Also, coagulopathy can be contributed by impaired wound healing or impaired clot formation.

Glanzmann's thrombasthenia is an abnormality of the platelets. It is an extremely rare coagulopathy, in which the platelets contain defective or low levels of glycoprotein IIb/IIIa (GpIIb/IIIa), which is a receptor for fibrinogen. As a result, no fibrinogen bridging of platelets to other platelets can occur, and the bleeding time is significantly prolonged.

Purpura is a condition of red or purple discolored spots on the skin that do not blanch on applying pressure. The spots are caused by bleeding underneath the skin secondary to platelet disorders, vascular disorders, coagulation disorders, or other causes. They measure 3–10 mm, whereas petechiae measure less than 3 mm, and ecchymoses greater than 1 cm.
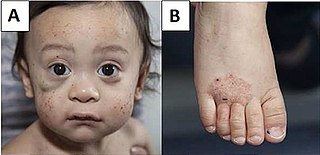
Wiskott–Aldrich syndrome (WAS) is a rare X-linked recessive disease characterized by eczema, thrombocytopenia, immune deficiency, and bloody diarrhea. It is also sometimes called the eczema-thrombocytopenia-immunodeficiency syndrome in keeping with Aldrich's original description in 1954. The WAS-related disorders of X-linked thrombocytopenia (XLT) and X-linked congenital neutropenia (XLN) may present with similar but less severe symptoms and are caused by mutations of the same gene.
Bernard–Soulier syndrome (BSS) is a rare autosomal recessive bleeding disorder that is caused by a deficiency of the glycoprotein Ib-IX-V complex (GPIb-IX-V), the receptor for von Willebrand factor. The incidence of BSS is estimated to be less than 1 case per million persons, based on cases reported from Europe, North America, and Japan. BSS is a giant platelet disorder, meaning that it is characterized by abnormally large platelets.
Mean platelet volume (MPV) is a machine-calculated measurement of the average size of platelets found in blood and is typically included in blood tests as part of the CBC. Since the average platelet size is larger when the body is producing increased numbers of platelets, the MPV test results can be used to make inferences about platelet production in bone marrow or platelet destruction problems.

Kasabach–Merritt syndrome, also known as hemangioma with thrombocytopenia, is a rare disease, usually of infants, in which a vascular tumor leads to decreased platelet counts and sometimes other bleeding problems, which can be life-threatening. It is also known as hemangioma thrombocytopenia syndrome. It is named after Haig Haigouni Kasabach and Katharine Krom Merritt, the two pediatricians who first described the condition in 1940.
Gray platelet syndrome (GPS), or platelet alpha-granule deficiency, is a rare congenital autosomal recessive bleeding disorder caused by a reduction or absence of alpha-granules in blood platelets, and the release of proteins normally contained in these granules into the marrow, causing myelofibrosis. The name derives from the initial observation of gray appearance of platelets with a paucity of granules on blood films from a patient with a lifelong bleeding disorder.

May–Hegglin anomaly (MHA), is a rare genetic disorder of the blood platelets that causes them to be abnormally large.
Hematologic diseases are disorders which primarily affect the blood & blood-forming organs. Hematologic diseases include rare genetic disorders, anemia, HIV, sickle cell disease & complications from chemotherapy or transfusions.
Congenital amegakaryocytic thrombocytopenia (CAMT) is a rare inherited disorder caused by mutations in the c-mpl gene. It is often diagnosed in the neonatal period due to petechia. It begins with congenital thrombocytopenia and can progress to aplastic anemia. It is associated with leukemia.
Harris platelet syndrome (HPS) is the most common inherited giant platelet disorder.

X-linked thrombocytopenia, also referred to as XLT or thrombocytopenia 1, is an inherited clotting disorder that primarily affects males. It is a WAS-related disorder, meaning it is caused by a mutation in the Wiskott–Aldrich syndrome (WAS) gene, which is located on the short arm of the X chromosome. WAS-related disorders include Wiskott–Aldrich syndrome, XLT, and X-linked congenital neutropenia (XLN). Of the WAS-related disorders, X-linked thrombocytopenia is considered to be the milder phenotype. Between 1 and 10 per million males worldwide are affected with this disorder. Females may be affected with this disorder but this is very rare since females have two X chromosomes and are therefore typically carriers of the mutation.

Epstein syndrome is a rare genetic disease characterized by a mutation in the MYH9 gene in nonmuscle myosin. This disease affects the patient's renal system and can result in kidney failure. Epstein Syndrome was first discovered in 1972 when two families had similar symptoms to Alport syndrome. Epstein syndrome and other Alport-like disorders were seen to be caused by mutations in the MYH9 gene, however, Epstein syndrome differs as it was more specifically linked to a mutation on the R702 codon on the MYH9 gene. Diseases with mutations on the MYH9 gene also include May–Hegglin anomaly, Sebastian syndrome and Fechtner syndrome.
