Epstein syndrome is a rare genetic disease characterized by a mutation in the MYH9 gene in nonmuscle myosin. This disease affects the patient's renal system and can result in kidney failure. Epstein syndrome was first discovered in 1972 when two families had similar symptoms to Alport syndrome.[1] Epstein syndrome and other Alport-like disorders were seen to be caused by mutations in the MYH9 (myosin heavy chain 9) [2] gene, however, Epstein syndrome differs as it was more specifically linked to a mutation on the R702 codon on the MYH9 gene. Diseases with mutations on the MYH9 gene also include May–Hegglin anomaly, Sebastian syndrome and Fechtner syndrome.[3]
Initial symptoms are often described as bleeding tendency and thrombocytopenia. Bleeding tendency may be observed in epistaxis and purpura.[4] Other symptoms may include macrothrombocytopenia, proteinuria, nephropathy, sensorineural hearing loss, low platelet count, oral lesions and cataracts. The most common symptoms include macrothrombocytopenia, mild sensorineural hearing loss and nephritis.[3] The symptoms and the severity of these symptoms vary between patients where most patients experience nephritis in childhood and then progress to kidney failure in adolescence.[5] In macrothrombocytopenia platelet sizes can reach to approximately 6.6um compared to a normal platelet size of 2.5um where 30% platelets can reach the size of an erythrocytes. This large platelet size can be compared with MYH9 disorders where platelet size can vary between 4.5um and the 6.6um that is found in Epstein syndrome with mutations on the R207 codon.[6][7]
Causes
DNA strand
Epstein syndrome is caused by a mutation in MYH9 gene.[6] This mutation is autosomal dominant and is thereby inherited if one or both parents carry the mutated gene. However, there have been cases where Epstein syndrome has been sporadic or non-congenital.[8] Mutations are found in the nonmuscle myosin heavy chain IIA and is associated with chromosome 22. The MYH9 gene encodes for tissues including platelets, cochlea, renal cells, neutrophils and eyes.[4]
Pathophysiology
Kidney tubules
The main symptom in Epstein syndrome is thrombocytopaenia. Thrombocytopaenia is generally inherited as an autosomal dominant gene and platelets are found to aggregate with either epinephrine or collagen. Platelets assist in blood clotting and coagulate when there are damages blood vessels. This coagulation attempts to cease bleeding. Thrombocytopaenia means there is low platelet volume in the blood. This means there are less platelets to coagulate in presence of a damaged blood vessel, which can result in bleeding problems.[9] The platelets are large (macrothrombocytopenia) and often consist of neutrophil inclusions.[10] The large size of platelets may be due to excess microtubule coils and tublin. This large size of the platelets also affects their ability to bind to each other to seal damaged blood vessels and stop bleeding.[11]
Nephritis involves the inflammation of the kidney and this inflammation results in a reduced ability to filter blood and remove nitrogenous wastes. This excessive accumulation of wastes will result in a continued build-up of wastes including urea which interferes with metabolism.[12]
One of the kidney's functions include osmoregulation which involves the regulation of osmotic pressure in the blood by regulating the water content in blood pressure. Through osmosis (water movement from a low solute concentration to an area of high solute concentration) water is reabsorbed back into the blood from nephron tubules in the kidney. This maintenance of water concentration in the blood is essential for maintaining blood pressure and is affected in nephritis.[12]
Diagnosis
The cardinal symptom in Epstein syndrome is thrombocytopaenia with giant platelets (macrothrombocytopenia). A peripheral blood smear is taken from the patient and a light microscope is used to these identify giant platelets. Leukocyte inclusions are also examined for, because approximately 41.1% of R207 mutations have leukocyte inclusions. These are often abnormal neutrophils with a small size of approximately less than 0.7um and have an abnormal[citation needed]
location on the non-muscle myosin heavy chain IIA (NMMHC-II). These inclusion bodies have RNA and no DNA.[13] RNA in these inclusion bodies can be located in immunofluorescence analysis with the anti nonmuscle heavy chain IIA antibody.[7]
Hematology analysers or a hemocytometer can be used to determine the amount of platelets. A sample of blood is drawn from a patient's arm. A small amount of platelets in blood smears compared to the normal range of 150,000 to 450,000 platelets in microliter of blood suggest thrombocytopaenia, which is a common symptom in Epstein syndrome.[9]
A urine sample is often collected where a urinalysis can be used to determine the volume of proteins excreted in urine. Abnormal amounts of protein detected means the patient has proteinuria. Patients with Epstein syndrome often have large proteinuria where they excrete above 3.5g of protein in their urine in a day. This is one of the initial signs of renal disease.[14]
Easy bruising and abnormal bleeding tendencies are also described in initial diagnosis.[5] This supports the common misdiagnosis for chronic idiopathic thrombocytopaenia purpura in patients with Epstein syndrome. These symptoms are often noticed in early childhood due to the congenital cause of the disease.[15]
Treatment
Epstein syndrome is regarded as a refractory disease. Treatments include renal transplantation, however, this may become problematic as patient's low platelet count (thrombocytopaenia) increase the risks of complications in surgery.[16] Successful Renal transplants can arise from cadavers or from live donors.[11] To minimize the risk of patient's losing too much blood during the perioperative period, HLA-matched platelet infusions can be used to maintain satisfactory platelet levels.[14] Nephritis involves the inflammation of the kidney and this inflammation results in a reduced ability to filter blood.[12] In order to ensure the transplanted kidney is recognised as 'self' by Major Histocompatibility cells, immunosuppression drugs are used post operation. Immunosuppression medication may include calcineurin inhibitor, antimetabolite and methylprednisolone and assist in suppressing the immune system's response to the transplanted kidney[14]
Maintenance
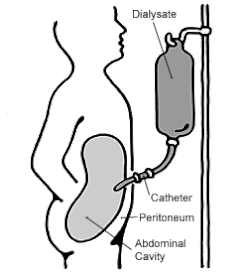
Peritoneal dialysis.
As a result of nephritis, a healthy blood pressure becomes difficult to maintain and hence medication including vasopressin may be prescribed to maintain blood pressure. Severe nephritis may mean kidney dialysis is required to ensure the blood is being filtered. This may include either peritoneal dialysis or haemodialysis, however, haemodialysis is most common as Epstein syndrome patients will often eventually have renal transplants.[11] Peritoneal dialysis involves blood being filtered through a membrane in the abdomen (peritoneum). Whereas haemodialysis involves blood being filtered through a dialyser, which consists of a semi-permeable membrane where toxins including urea are removed from the blood. Haemodialysis also filters blood quicker than peritoneal dialysis.[17]
Intravenous immunoglobin treatments can be used to support the patient's immune system as well as medication with Prednisolone to reduce inflammation.[18]
Sensorineural hearing loss is a common symptom in Epstein syndrome and can be treated with cochlea implants.[4] Cochlea implants have four main parts including the electrode array, the transmitter, the receiver/stimulator and the microphone. The microphone is positioned behind the ear and receives sound waves, the processor then converts the sound into electrical signals. These electrical signals are further converted into electrical impulses in the cochlea where the transmitter is located, which in turn sends the signal to the auditory nerve. This results in a nervous impulse sent to the brain where the 'sound' is deciphered.[19]
Prognosis
In most case Epstein syndrome patients will endure early-onset end-stage renal disease (ESRD) at the end of adolescence. Expected kidney failure means patients will need renal transplants in the near future.[16]
Notable cases
In a study which investigated suitable management for severe Epstein syndrome, four patients were observed. Patient 1 demonstrated a common background and symptoms for Epstein syndrome. Patient 1 was a male who was initially diagnosed with chronic idiopathic thrombocytopaenia purpura in early childhood and then macrothrombocytopaenia. He progressed to kidney failure and began haemodialysis at seventeen years old. Patient 1 was only diagnosed with Epstein syndrome at the age of thirty-three where the mutation in the nonmuscle myosin was noticed. His eldest son also carried the same mutation.[15]
An Epstein syndrome patient with oral lesions was recorded to be the first with this symptom. The patient was a twenty-six-year-old female who was diagnosed with thrombocytopaenia with purpura at age four. She was then finally diagnosed with Epstein syndrome at twenty-two years old when she developed symptoms for hearing loss as well as proteinuria and hematuria revealing poor kidney function. After an oral examination, palatal purpura in her mouth was observed.[20]
A Japanese male was diagnosed with sporadic Epstein syndrome where both his parents did not have the R702H mutation. He was initially diagnosed with chronic macrothrombocytopaenia at three years old and throughout childhood symptoms for hearing loss and proteinuria were observed. At 24 years old his kidney failure had severely progressed as well as his hearing loss and thrombocytopenia. A genetic test confirmed he had the R702H mutation despite having no familial record of Epstein syndrome.[8]
↑ Hao, Jihong; Kada, Akiko; Kunishima, Shinji (2018). "Further classification of neutrophil non-muscle myosin heavy chain-IIA localization for efficient genetic diagnosis of MYH9 disorders". Annals of Hematology. 97 (4): 709–711. doi:10.1007/s00277-017-3195-3. PMID29199357. S2CID3647689.
1 2 3 Hashimoto, Junya (2015). "Successful Kidney Transplantation in Epstein Syndrome With Antiplatelet Antibodies and Donor-specific Antibodies: A Case Report". Transplantation Proceedings. 47 (8): 2541–2543. doi:10.1016/j.transproceed.2015.09.010. PMID26518967.
1 2 Hashimoto, Junya (2018). "Management of patients with severe Epstein syndrome: Review of four patients who received living-donor renal transplantation". Nephrology. 24 (4): 450–455. doi:10.1111/nep.13253. PMID29532554. S2CID3873853.
1 2 Hamasaki, Yuko (2018). "Management of patients with severe Epstein syndrome: Review of four patients who received living-donor renal transplantation'". Nephrology. 24 (4): 450–455. doi:10.1111/nep.13253. PMID29532554. S2CID3873853.
↑ Richards (1991). "Epstein syndrome: oral lesions ina patient with nephropathy,deafness and thrombocytopenia". Journal of Oral Pathology and Medicine. 20 (10): 512–513. doi:10.1111/j.1600-0714.1991.tb00415.x. PMID1753355.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.