Hemolysis or haemolysis, also known by several other names, is the rupturing (lysis) of red blood cells (erythrocytes) and the release of their contents (cytoplasm) into surrounding fluid. Hemolysis may occur in vivo or in vitro.

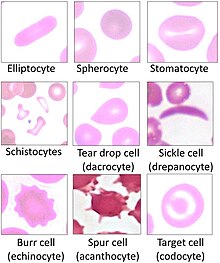
Spherocytosis is the presence of spherocytes in the blood, i.e. erythrocytes that are sphere-shaped rather than bi-concave disk shaped as normal. Spherocytes are found in all hemolytic anemias to some degree. Hereditary spherocytosis and autoimmune hemolytic anemia are characterized by having only spherocytes.
Hereditary spherocytosis (HS) is a congenital hemolytic disorder wherein a genetic mutation coding for a structural membrane protein phenotype causes the red blood cells to be sphere-shaped (spherocytosis), rather than the normal biconcave disk shape. This abnormal shape interferes with the cells' ability to flex during blood circulation, and also makes them more prone to rupture under osmotic stress, mechanical stress, or both. Cells with the dysfunctional proteins are degraded in the spleen, which leads to a shortage of erythrocytes and results in hemolytic anemia.

Hemolytic anemia or haemolytic anaemia is a form of anemia due to hemolysis, the abnormal breakdown of red blood cells (RBCs), either in the blood vessels or elsewhere in the human body (extravascular). This most commonly occurs within the spleen, but also can occur in the reticuloendothelial system or mechanically. Hemolytic anemia accounts for 5% of all existing anemias. It has numerous possible consequences, ranging from general symptoms to life-threatening systemic effects. The general classification of hemolytic anemia is either intrinsic or extrinsic. Treatment depends on the type and cause of the hemolytic anemia.

Pyruvate kinase deficiency is an inherited metabolic disorder of the enzyme pyruvate kinase which affects the survival of red blood cells. Both autosomal dominant and recessive inheritance have been observed with the disorder; classically, and more commonly, the inheritance is autosomal recessive. Pyruvate kinase deficiency is the second most common cause of enzyme-deficient hemolytic anemia, following G6PD deficiency.
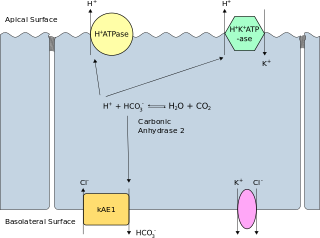
Band 3 anion transport protein, also known as anion exchanger 1 (AE1) or band 3 or solute carrier family 4 member 1 (SLC4A1), is a protein that is encoded by the SLC4A1 gene in humans.

Hereditary pyropoikilocytosis (HPP) is an autosomal recessive form of hemolytic anemia characterized by an abnormal sensitivity of red blood cells to heat and erythrocyte morphology similar to that seen in thermal burns or from prolonged exposure of a healthy patient's blood sample to high ambient temperatures. Patients with HPP tend to experience severe hemolysis and anemia in infancy that gradually improves, evolving toward typical elliptocytosis later in life. However, the hemolysis can lead to rapid sequestration and destruction of red cells. Splenectomy is curative when this occurs.
Cold agglutinin disease (CAD) is a rare autoimmune disease characterized by the presence of high concentrations of circulating cold sensitive antibodies, usually IgM and autoantibodies that are also active at temperatures below 30 °C (86 °F), directed against red blood cells, causing them to agglutinate and undergo lysis. It is a form of autoimmune hemolytic anemia, specifically one in which antibodies bind red blood cells only at low body temperatures, typically 28–31 °C.

Ankyrins are a family of proteins that mediate the attachment of integral membrane proteins to the spectrin-actin based membrane cytoskeleton. Ankyrins have binding sites for the beta subunit of spectrin and at least 12 families of integral membrane proteins. This linkage is required to maintain the integrity of the plasma membranes and to anchor specific ion channels, ion exchangers and ion transporters in the plasma membrane. The name is derived from the Greek word ἄγκυρα (ankyra) for "anchor".

Protein 4.1,, is a protein associated with the cytoskeleton that in humans is encoded by the EPB41 gene. Protein 4.1 is a major structural element of the erythrocyte membrane skeleton. It plays a key role in regulating membrane physical properties of mechanical stability and deformability by stabilizing spectrin-actin interaction. Protein 4.1 interacts with spectrin and short actin filaments to form the erythrocyte membrane skeleton. Mutations of spectrin and protein 4.1 are associated with elliptocytosis or spherocytosis and anemia of varying severity.

Erythrocyte membrane protein band 4.2 is a protein that in humans is encoded by the EPB42 gene. It is part of the red blood cell cytoskeleton.

Aldolase A deficiency is an autosomal recessive metabolic disorder resulting in a deficiency of the enzyme aldolase A; the enzyme is found predominantly in red blood cells and muscle tissue. The deficiency may lead to hemolytic anaemia as well as myopathy associated with exercise intolerance and rhabdomyolysis in some cases.

Spectrin alpha chain, erythrocyte is a protein that in humans is encoded by the SPTA1 gene.
Hexokinase deficiency is an extremely rare autosomal recessive condition that falls under the category of erythroenzymopathies, or defects in red cell enzymes. Hexokinase deficiency manifests is associated with chronic nonspherocytic hemolytic anemia. Hemolytic anemia seems to be the only clinical sign of hexokinase deficiency. In 1967 the first case of hexokinase deficiency was described by Valentine et al, since then, less than 50 cases have been reported.
Congenital hemolytic anemia (CHA) is a diverse group of rare hereditary conditions marked by decreased life expectancy and premature removal of erythrocytes from blood flow. Defects in erythrocyte membrane proteins and red cell enzyme metabolism, as well as changes at the level of erythrocyte precursors, lead to impaired bone marrow erythropoiesis. CAH is distinguished by variable anemia, chronic extravascular hemolysis, decreased erythrocyte life span, splenomegaly, jaundice, biliary lithiasis, and iron overload. Immune-mediated mechanisms may play a role in the pathogenesis of these uncommon diseases, despite the paucity of data regarding the immune system's involvement in CHAs.
Mechanical hemolytic anemia is a form of hemolytic anemia due to mechanically induced damage to red blood cells. Red blood cells, while flexible, may in some circumstances succumb to physical shear and compression. This may result in hemoglobinuria. The damage is induced through repetitive mechanical motions such as prolonged marching and marathon running. Mechanical damage can also be induced through the chronic condition microangiopathic hemolytic anemia or due to prosthetic heart valves.
Erythrocyte fragility refers to the propensity of erythrocytes to hemolyse (rupture) under stress. It can be thought of as the degree or proportion of hemolysis that occurs when a sample of red blood cells are subjected to stress. Depending on the application as well as the kind of fragility involved, the amount of stress applied and/or the significance of the resultant hemolysis may vary.

Stomatin also known as human erythrocyte integral membrane protein band 7 is a protein that in humans is encoded by the STOM gene.

PIEZO1 is a mechanosensitive ion channel protein that in humans is encoded by the gene PIEZO1. PIEZO1 and its close homolog PIEZO2 were cloned in 2010, using an siRNA-based screen for mechanosensitive ion channels.
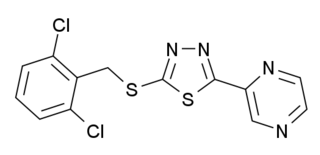
Yoda1 is a chemical compound which is the first agonist developed for the mechanosensitive ion channel PIEZO1. This protein is involved in regulation of blood pressure and red blood cell volume, and Yoda1 is used in scientific research in these areas.