
Jaundice, also known as icterus, is a yellowish or greenish pigmentation of the skin and sclera due to high bilirubin levels. Jaundice in adults is typically a sign indicating the presence of underlying diseases involving abnormal heme metabolism, liver dysfunction, or biliary-tract obstruction. The prevalence of jaundice in adults is rare, while jaundice in babies is common, with an estimated 80% affected during their first week of life. The most commonly associated symptoms of jaundice are itchiness, pale feces, and dark urine.

Caroli disease is a rare inherited disorder characterized by cystic dilatation of the bile ducts within the liver. There are two patterns of Caroli disease: focal or simple Caroli disease consists of abnormally widened bile ducts affecting an isolated portion of liver. The second form is more diffuse, and when associated with portal hypertension and congenital hepatic fibrosis, is often referred to as "Caroli syndrome". The underlying differences between the two types are not well understood. Caroli disease is also associated with liver failure and polycystic kidney disease. The disease affects about one in 1,000,000 people, with more reported cases of Caroli syndrome than of Caroli disease.

Biliary atresia, also known as extrahepatic ductopenia and progressive obliterative cholangiopathy, is a childhood disease of the liver in which one or more bile ducts are abnormally narrow, blocked, or absent. It can be congenital or acquired. It has an incidence of one in 10,000–15,000 live births in the United States, and a prevalence of one in 16,700 in the British Isles. Biliary atresia is most common in East Asia, with a frequency of one in 5,000.

Primary biliary cholangitis (PBC), previously known as primary biliary cirrhosis, is an autoimmune disease of the liver. It results from a slow, progressive destruction of the small bile ducts of the liver, causing bile and other toxins to build up in the liver, a condition called cholestasis. Further slow damage to the liver tissue can lead to scarring, fibrosis, and eventually cirrhosis.

Hep G2 is a human liver cancer cell line.
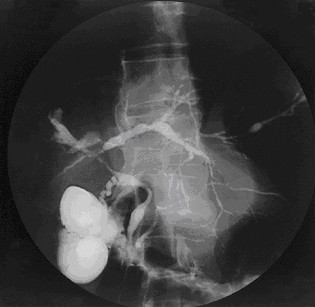
Primary sclerosing cholangitis (PSC) is a long-term progressive disease of the liver and gallbladder characterized by inflammation and scarring of the bile ducts, which normally allow bile to drain from the gallbladder. Affected individuals may have no symptoms or may experience signs and symptoms of liver disease, such as yellow discoloration of the skin and eyes, itching, and abdominal pain.

Aagenaes syndrome is a syndrome characterised by congenital hypoplasia of lymph vessels, which causes lymphedema of the legs and recurrent cholestasis in infancy, and slow progress to hepatic cirrhosis and giant cell hepatitis with fibrosis of the portal tracts.

Alagille syndrome (ALGS) is a genetic disorder that affects primarily the liver and the heart. Problems associated with the disorder generally become evident in infancy or early childhood. The disorder is inherited in an autosomal dominant pattern, and the estimated prevalence of Alagille syndrome is 1 in every 30,000 to 1 in every 40,000 live births. It is named after the French pediatrician Daniel Alagille, who first described the condition in 1969. Children with Alagille syndrome live to the age of 18 in about 90% of the cases.

Citrullinemia is an autosomal recessive urea cycle disorder that causes ammonia and other toxic substances to accumulate in the blood.
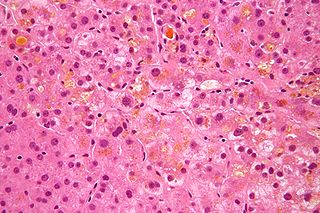
Cholestasis is a condition where the flow of bile from the liver to the duodenum is impaired. The two basic distinctions are:

Dubin–Johnson syndrome is a rare, autosomal recessive, benign disorder that causes an isolated increase of conjugated bilirubin in the serum. Classically, the condition causes a black liver due to the deposition of a pigment similar to melanin. This condition is associated with a defect in the ability of hepatocytes to secrete conjugated bilirubin into the bile, and is similar to Rotor syndrome. It is usually asymptomatic, but may be diagnosed in early infancy based on laboratory tests. No treatment is usually needed.
Rotor syndrome is a rare cause of mixed direct (conjugated) and indirect (unconjugated) hyperbilirubinemia, relatively benign, autosomal recessive bilirubin disorder characterized by non-hemolytic jaundice due to the chronic elevation of predominantly conjugated bilirubin.
Neonatal cholestasis refers to elevated levels of conjugated bilirubin identified in newborn infants within the first few months of life. Conjugated hyperbilirubinemia is clinically defined as >20% of total serum bilirubin or conjugated bilirubin concentration greater than 1.0 mg/dL regardless of total serum bilirubin concentration. The differential diagnosis for neonatal cholestasis can vary extensively. However, the underlying disease pathology is caused by improper transport and/or defects in excretion of bile from hepatocytes leading to an accumulation of conjugated bilirubin in the body. Generally, symptoms associated with neonatal cholestasis can vary based on the underlying cause of the disease. However, most infants affected will present with jaundice, scleral icterus, failure to thrive, acholic or pale stools, and dark urine.
Intrahepatic cholestasis of pregnancy (ICP), also known as obstetric cholestasis, cholestasis of pregnancy, jaundice of pregnancy, and prurigo gravidarum, is a medical condition in which cholestasis occurs during pregnancy. It typically presents with itching and can lead to complications for both mother and fetus.

Liver cancer, also known as hepatic cancer, primary hepatic cancer, or primary hepatic malignancy, is cancer that starts in the liver. Liver cancer can be primary in which the cancer starts in the liver, or it can be liver metastasis, or secondary, in which the cancer spreads from elsewhere in the body to the liver. Liver metastasis is the more common of the two liver cancers. Instances of liver cancer are increasing globally.

ATP-binding cassette, sub-family B member 11 (ABCB11), also known as the bile salt export pump (BSEP), is a protein which in humans is encoded by the ABCB11 gene.

The ATP-binding cassette 4 (ABCB4) gene encodes multidrug resistance protein 3. ABCB4 is associated with progressive familial intrahepatic cholestasis type 3 and intrahepatic cholestasis of pregnancy.

Probable phospholipid-transporting ATPase IC is an enzyme that in humans is encoded by the ATP8B1 gene. This protein is associated with progressive familial intrahepatic cholestasis type 1 as well as benign recurrent intrahepatic cholestasis.

Odevixibat, sold under the brand name Bylvay among others, is a medication for the treatment of progressive familial intrahepatic cholestasis. It is taken by mouth. Odevixibat is a reversible, potent, selective inhibitor of the ileal bile acid transporter (IBAT). It was developed by Albireo Pharma.
Bile acid synthesis disorders (BASDs) are rare metabolic disorders characterized by defects in the synthesis of bile acids, which are crucial for cholesterol breakdown and the absorption of fats and fat-soluble vitamins. These disorders can lead to the accumulation of abnormal bile acids and intermediary metabolites, causing damage to various organs.
