obstructive type of cholestasis, where there is a mechanical blockage in the duct system that can occur from a gallstone or malignancy, and
metabolic type of cholestasis, in which there are disturbances in bile formation that can occur because of genetic defects or acquired as a side effect of many medications.
Classification is further divided into acute or chronic and extrahepatic or intrahepatic.
Signs and symptoms
The signs and symptoms of cholestasis vary according to the cause. In case of sudden onset, the disease is likely to be acute, while the gradual appearance of symptoms suggests chronic pathology.[2] In many cases, patients may experience pain in the abdominal area. Localization of pain to the upper right quadrant can be indicative of cholecystitis or choledocholithiasis, which can progress to cholestasis.[3][4]
Pruritus or itching is often present in many patients with cholestasis.[5] Patients may present with visible scratch marks as a result of scratching.[2] Pruritus is often misdiagnosed as a dermatological condition, especially in patients that do not have jaundice as an accompanying symptom.[2] In a typical day, pruritus worsens as the day progresses, particularly during the evening time.[6] Overnight, pruritus dramatically improves. This cycle can be attributed to an increase in the concentration of biliary elements during the day due to food consumption, and a decline at night.[2] Pruritus is mostly localized to the limbs, but can also be more generalized.[6] The efficacy of naltrexone for cholestatic pruritus suggests involvement of the endogenous opioid system.
Many patients may experience jaundice as a result of cholestasis.[7] This is usually evident after physical examination as yellow pigment deposits on the skin, in the oral mucosa, or conjunctiva.[2][8] Jaundice is an uncommon occurrence in intrahepatic (metabolic) cholestasis, but is common in obstructive cholestasis. The majority of patients with chronic cholestasis also experience fatigue.[9] This is likely a result of defects in the corticotrophin hormone axis or other abnormalities with neurotransmission.[2] Some patients may also have xanthomas, which are fat deposits that accumulate below the skin.[10] These usually appear waxy and yellow, predominantly around the eyes and joints.[11] This condition results from an accumulation of lipids within the blood.[12] If gallstones prevent bile flowing from the pancreas to the small intestine, it can lead to gallstone pancreatitis. Physical symptoms include nausea, vomiting, and abdominal pain.
Bile is required for the absorption of fat-soluble vitamins.[13] As such, patients with cholestasis may present with a deficiency in vitamins A, D, E, or K due to a decline in bile flow.[14] Patients with cholestasis may also experience pale stool and dark urine.[15]
Causes
Relative incidences of causes of neonatal cholestasis (data from Germany)
Acute and chronic cholestasis can be caused by certain drugs or their metabolites. Drug-induced cholestasis (DIC) falls under drug-induced liver injury (DILI), specifically the cholestatic or mixed type.[23][24] While some drugs (e.g., acetaminophen) are known to cause DILI in a predictable dose-dependent manner (intrinsic DILI), most cases of DILI are idiosyncratic, i.e., affecting only a minority of individuals taking the medication.[25][26] Seventy-three percent of DIC cases can be attributed to a single prescription medication, commonly antibiotics and antifungals, anti-diabetics, anti-inflammatory, and cardiovascular drugs, psychotropic drugs.[24][27] The exact pathomechanism may vary for different drugs and requires further elucidation.[27]
Typical symptoms of DIC include pruritus and jaundice, nausea, fatigue, and dark urine, which usually resolve after discontinuation of the offending medication.[24][28]
Antibiotics and antifungals that commonly cause DIC are penicillins, macrolides, trimethoprim/sulfamethoxazole, and tetracyclines.[29][32] Due to its clavulanic acid component, penicillin amoxicillin-clavulanate is the most common culprit of cholestatic liver injury.[29]Flucloxacillin, which is commonly prescribed in the UK, Sweden, and Australia, is another penicillin frequently implicated in DIC. Cholestasis induced by penicillins usually resolves after withdrawal.[29]Macrolides with cholestatic potential include erythromycin, clarithromycin, and azithromycin, and prognosis is likewise favorable with these drugs.[29]Trimethoprim/sulfamethoxazole (via its sulfonamide component) is the fourth most common antibiotic responsible for DILI in North America. However, DIC is comparatively less common with low-dose tetracyclines like doxycycline.[29] Other cholestatic antimicrobials include the antifungal terbinafine, notable for its potential to cause life-threatening cholestatic injury, and quinolones (ciprofloxacin, levofloxacin), which have been linked to cholestatic hepatitis and vanishing bile duct syndrome.[29]
Among psychotropic drugs, chlorpromazine is known to cause cholestatic hepatitis. Tricyclic antidepressants (imipramine, amitriptyline) and SSRIs (duloxetine) causing cholestasis have also been reported.[29] Anti-inflammatory drugs with cholestatic potential include the immunosuppressant azathioprine, which has been reported to cause fatal cholestatic hepatitis, and the NSAID diclofenac.[29]
Chronic cholestasis occurs in primary biliary cholangitis (PBC). PBC is a progressive autoimmune liver disease in which small intrahepatic bile ducts are selectively destroyed, leading to cholestasis, biliary fibrosis, cirrhosis, and eventually liver failure that requires transplantation.[33][34][35] Prevalence of PBC ranges from 19 to 402 cases/million depending on geographic location,[33] with a 9:1 female preponderance[36] and median ages of diagnosis of 68.5 years for females and 54.5 years for males.[37]
At diagnosis, 50% of PBC patients are asymptomatic, indicative of an early stage of disease, while another 50% report fatigue and daytime sleepiness. Other symptoms include pruritus and skin lesions, and in prolonged cholestasis, malabsorption and steatorrhea leading to fat-soluble vitamin deficiency. Disease progression is accompanied by intensifying portal hypertension and hepatosplenomegaly. Clinically, diagnosis generally requires a 1:40 or greater titer of anti-mitochondrial antibody (AMA) against PDC-E2 and elevated alkaline phosphatase persisting for 6+ months.[33]
Ursodeoxycholic acid (UDCA) is an FDA-approved first-line treatment for PBC. At moderate doses, UDCA has been demonstrated to slow disease progression and improve transplant-free survival. A complete response is achieved in 25–30% patients, and similar survival as the general population is expected in 2/3 of patients on UDCA. For the 1/3 non-responders, obeticholic acid (OCA) is approved by the FDA as a second-line treatment.[33]
The precise etiology of PBC remains poorly understood, though a clearer picture is starting to emerge. A loss of immune tolerance is indicated by the presence of AMAs and autoreactive CD4+ and CD8+ T cells targeting cholangiocytes that line the bile ducts.[34] Cholangiocytes are normally responsible for 40% of bile flow, mostly through secretion of bicarbonate into bile via anion exchanger 2 (AE2) on their apical membrane.[35] The resulting bicarbonate "umbrella" that forms over cholangiocytes provides protection from toxic bile salts.[35] However, in PBC there is repression of AE2 activity due to upregulation of miR-506. This results in decreased biliary bicarbonate secretion and consequently, cholestasis and injury to cholangiocytes by bile salts.[35][38] Injury may induce cholangiocytes to undergo apoptosis, and during this process, the unique way in which cholangiocytes handle the degradation of PDC-E2 (the E2 subunit of mitochondrial pyruvate dehydrogenase complex) may be a trigger for PSC. Specifically, PDC-E2 in apoptotic cholangiocytes undergo a covalent modification that may render them recognizable to antibodies and thereby trigger a break in self-tolerance.[35] The problem is compounded by cholangiocytes' peculiarly abundant expression of HLA-II and HLA-I, as well as adhesion and chemoattractant molecules, which recruit aid in recruitment of mononuclear immune cells.[39][35]
Both genetic and environmental factors probably contribute to PBC pathogenesis.[34] Genetic predisposition is suggested by high concordance between identical twins, higher incidence among relatives, and a strong association of disease with certain HLA variants.[34] Disease is likely triggered in the genetically predisposed by some environmental factor, such as pollutants, xenobiotics (e.g., chemicals in makeup), diet, drugs, stress, and infectious agents. Urinary tract infection with E. coli is a particularly strong risk factor for PBC. A possible explanation is that E. coli possess a similar PDC-E2 as humans which could trigger autoimmunity via molecular mimicry.[35][39][34]
Chronic cholestasis is a feature in primary sclerosing cholangitis (PSC). PSC is a rare and progressive cholestatic liver disease characterized by narrowing, fibrosis, and inflammation of intrahepatic or extrahepatic bile ducts, leading to reduced bile flow or formation (i.e., cholestasis).[40][41] The pathogenesis of PSC remains unclear but probably involves a combination of environmental factors and genetic predisposition.[41] Notably, 70-80% of patients with PSC are comorbid with inflammatory bowel disease (e.g., ulcerative colitis or Crohn's colitis), suggesting there exists a link between the two.[40][42]
PSC predominantly affects males (60–70%) of 30–40 years of age.[41] The disease has an incidence is 0.4–2.0 cases/100,000 and a prevalence of 16.2 cases/100,000, making it a rare disease.[43][41] Nonetheless, PSC accounts for 6% of liver transplants in the US due to its eventual progression to end-stage liver disease, with a mean transplant-free survival of 21.3 year.[41]
Diagnosis requires elevated serum alkaline phosphatase persisting for at least 6 months and the presence of bile duct strictures on cholangiogram.[40][41] Unlike primary biliary cholangitis, PSC lacks a diagnostic autoantibody or reliable biomarker of disease progression.[41][40] Although a liver biopsy is not required for diagnosis, the characteristic histological finding is concentric periductal fibrosis resembling onion skin.[41]
PSC is associated with increased risk of several cancers, most notably, a 400 times greater risk for cholangiocarcinoma compared to the general population.[40] Patients with PSC also face elevated risk of pancreatic and colorectal cancer.[41] Therefore, regular screening is recommended.[40]
No drugs are currently approved for treating PSC specifically.[33] Although commonly given, ursodeoxycholic acid at moderate doses failed to improve transplant-free survival in randomized controlled trials.[41][40] Due to disease progression, 40% of patients eventually require liver transplantation, which has survival rates (91% at 1 year, 82% at 5 years, and 74% at 10 years).[41] However, the disease recurs in at least 25% of transplant recipients, particularly in those with IBD and an intact colon.[40] Clinical trials are underway for several novel therapies, including obeticholic acid (a bile acid analogue), simtuzumab (a monoclonal antibody), and 24-norursodeoxycholic acid (a synthetic bile acid).[40]
Although the pathogenesis of PSC is poorly understood, three dominant theories have been proposed: 1) aberrant immune response, 2) increased intestinal permeability, and 3) dysbiosis of gut microbiota.[44] The first theory involves immune-mediated damage to bile ducts by T cells. In PSC, cholangiocytes and hepatocytes display aberrant expression of adhesion molecules, which facilitate homing of intestinal T cells to the liver.[44][40] Additionally, intestinal microbiota may produce pathogen-associated molecular patterns that stimulate cholangiocytes and hepatic macrophages to produce proinflammatory cytokines, which promote recruitment of immune cells to the bile ducts, fibrosis, cholangiocyte apoptosis and senescence, and ultimately destruction of the bile ducts.[40][35] In support of T cell involvement, certain human leukocyte antigen (HLA) variants are strongly associated with PSC risk.[40] Further evidence for genetic predisposition include the identification of 23 non-HLA susceptibility loci and a higher disease risk among siblings, though environmental factors appear to play a much greater role in pathogenesis.[42]
Another theory postulates that increased intestinal permeability contributes to PSC. Tight junctions, which normally maintain the integrity of the intestinal epithelium, may become disrupted in inflammation.[44] Leaky tight junctions could allow commensal bacteria and toxins to enter portal circulation and reach the liver, where they can trigger inflammation and fibrosis.[44]
The intestinal dysbiosis theory hypothesizes that yet unidentified environmental triggers (e.g., diet, medication, inflammation) reduce microbiota diversity and/or alter the population of specific species.[44] The resulting imbalance between primary and secondary bile acids may lead to PSC via the gut-liver axis.[44] The primary bile acids cholic acid (CA) and chenodeoxycholic acid (CDCA) are synthesized in the liver and undergo conjugation before being released into the small intestine to aid digestion.[42] In the distal ileum, 95% of these conjugated BAs are actively reabsorbed via ASBT but 5% enter the colon and are converted by gut microbes into deconjugated secondary bile acids, predominantly deoxycholic acid (DCA) and lithocholic acid (LCA).[45][42] DCA and LCA are then reabsorbed into portal circulation and reach the liver, where they serve as signaling molecules that maintain bile acid homeostasis.[42] Specifically, DCA and LCA and potent agonists of farnesoid X receptor (FXR) and Takeda G protein-coupled receptor 5 (TGR5),[42] both of which mediate anti-inflammatory and cholangioprotective effects upon activation.[44] On cholangiocytes, TGR5 activation induces CFTR to secrete chloride into bile ducts, which then drives anion exchanger 2 to secrete bicarbonate into bile canaliculi.[42]Bicarbonate serves to protect the apical surface of cholangiocytes from damage by bile acids.[42] On macrophages, activation of FXR and TGR5 inhibits NF-κB, thereby reducing production of proinflammatory cytokines.[42] Therefore, it is hypothesized that a reduction in secondary bile acid production, as a result of dysbiosis, could lead to bile duct damage via decreased activation of FXR and TGR5. Indeed, lower levels of secondary bile acids were found in PSC patients, but a causal relationship is yet to be confirmed.[46][47]
Familial intrahepatic cholestasis (FIH) is a group of disorders that lead to intrahepatic cholestasis in children.[48] Most often, FIH occurs during the first year of life, with an incidence rate of 1/50,000 to 1/100,000.[49] There are three different versions of FIH, with each causing a different severity of jaundice. Typically, children exhibit recurrent jaundice episodes, which eventually become permanent.[48] Diagnosis usually occurs by analyzing laboratory features, liver biopsy results, DNA/RNA sequences, and biliary lipid analysis.[48] The definitive treatment for FIH is liver transplant which usually results in a high recovery rate.[48] Each type of FIH is a result of a different mutation. The three genes thought to be involved include APT8B1, which encodes for the FIC1 protein.[50] The ABCB11 gene encodes for the bile salt export pump (BSEP) protein, and the ABCB4 gene encodes for the multidrug resistance 3 (MDR3) protein.[50][51] BSEP and MDR3 are respectively responsible for transporting bile salt and phospholipid, two major constituents of bile, across the apical membrane of hepatocytes.[52]
Alagille syndrome is an autosomal dominant disorder that impacts five systems, including the liver, heart, skeleton, face, and eyes.[48] In the early part of life (within the first three months), patients with Alagille syndrome exhibit conjugated hyperbilirubinemia, severe pruritus, and jaundice.[48] Bile duct obliteration usually worsens over time, causing cirrhosis of the liver and eventual failure.[48] Diagnosis usually occurs using the classic criteria by looking at changes associated with the five systems discussed earlier.[53] Like FIH, the definitive treatment is a liver transplant.[54] Almost all patients with Alagille syndrome have mutations of the genes involved in the Notch signaling pathway. Most have a mutation of the JAG1 gene, while a small minority have a mutation of the NOTCH2 gene.[55][56]
Sepsis
A variety of factors associated with sepsis may cause cholestasis. Typically, patients have conjugated hyperbilirubinemia and alkaline phosphatase (ALP) elevation but not to extreme levels.[57] Sepsis-induced cholestasis may occur due to increased serum lipopolysaccharide levels. Lipopolysaccharides can inhibit and down-regulate bile salt transporters in hepatocytes, thereby leading to cholestasis.[48] As such, in the case of sepsis, cholestasis occurs not as a result of impaired obstruction but rather the disruption of bile flow. Ischemic liver injury resulting from sepsis can also cause cholestasis. Importantly, jaundice is not indicative of cholestasis in all cases. Widespread hemolysis resulting from sepsis may release bilirubin, thereby overwhelming bilirubin reabsorption and excretion mechanism.[48]
TPN-based cholestasis
Total parenteral nutrition (TPN) is given to patients with intestinal failure or a variety of other gastrointestinal problems.[48] Under normal settings, TPN causes a slight elevation of ALP levels. However, this does not indicate cholestasis alone.[48] In the case of TPN-induced cholestasis, there is an excessive elevation of ALP, gamma-glutamyltransferase (GGT), and conjugated bilirubin.[58] Without appropriate intervention, symptoms can quickly exacerbate, leading to liver cirrhosis and failure.[48] Cholestasis arising from TPN has a diverse range of causes, including toxicity to TPN components, underlying disorders, or a lack of enteral nutrition.[48] Without enteral food consumption, gallbladder function is greatly inhibited, leading to gallstone formation, subsequent blockage, and eventually cholestasis.[48] Cholestasis resulting from TPN may also be a result of reduced bile flow from portal endotoxins.[48] With TPN, there is a reduction in gastrointestinal motility, immunity, with an increase in permeability.[48] These changes facilitate bacteria growth and increase the amount of circulating endotoxin. Moreover, given that patients using TPN often have underlying health problems, drugs being used with known liver toxicity may also cause cholestasis. Lipids in TPN may cause cholestasis and liver damage by overwhelming clearage mechanisms.[59] Intravenous glucose can also cause cholestasis as a result of increased fatty acid synthesis and decreased breakdown, which facilitates the accumulation of fats.[60]
Intrahepatic cholestasis of pregnancy (obstetric cholestasis)
Intrahepatic cholestasis of pregnancy (ICP) is an acute cause of cholestasis that manifests most commonly in the third trimester of pregnancy.[15] It affects 0.5–1.5% of pregnancies in Europe and the US and up to 28% in women of Mapuche ethnicity in Chile.[61] ICP is characterized by severe pruritus and elevated serum levels of bile acids as well as transaminases and alkaline phosphatase.[62] These signs and symptoms resolve on their own shortly after delivery, though they may reappear in subsequent pregnancies for 45–70% of women.[63] In the treatment of ICP, current evidence suggests ursodeoxycholic acid (UDCA), a minor secondary bile acid in humans, is the most effective drug for reducing pruritus and improving liver function.[62]
The etiology of ICP is multifactorial and likely involves hormonal, genetic, and environmental factors. Several observations suggest estrogen plays a major role: ICP begins in the third trimester, when estrogen levels are highest, resolves after estrogen levels return to normal post-delivery, and occurs with higher incidence in multiple pregnancies, where estrogen levels are more elevated than usual.[63][64] Although estrogen's exact pathomechanism in ICP remains unclear, several explanations have been offered. Estrogen may induce a decrease in the fluidity of the hepatic sinusoidal membrane, leading to a decrease in the activity of basolateral Na+/K+-ATPase.[63][65][66] A weaker Na+ gradient results in diminished sodium-dependent uptake of bile acids from venous blood into hepatocytes by the sodium/bile acid cotransporter.[63][67] More recent evidence suggests that estrogen promotes cholestasis via its metabolite estradiol-17-β-D-glucuronide (E2).[61][63] E2 secreted into the canaliculi by MRP2 was found to repress the transcription of bile salt export pump (BSEP),[68][61][69] the apical ABC transporter responsible for exporting monoanionic conjugated bile acids from hepatocytes into bile canaliculi.[68] E2 was also found to upregulate miR-148a, which represses expression of the pregnane X receptor (PXR).[68] PXR is a nuclear receptor in hepatocytes that senses intracellular bile acid concentrations and regulates gene expression accordingly to increase bile efflux.[70]
Genetic predisposition for ICP is suggested by familial and regional clustering of cases.[63][61] Several studies have implicated heterozygous mutations of the genes ABCB11 and ABCB4 in ICP, which respectively encode the canalicular transport proteins BSEP and multidrug resistance protein 3 (MDR3).[71][61] MDR3 is responsible for exporting phosphatidylcholine, the major lipid component of bile, into bile canaliculi where it forms micelles with bile salts to prevent the latter from damaging luminal epithelium. Bile flow requires canalicular secretion of both bile salts and phosphatidylcholine.[71] MDR3 mutations are an established predisposing factor, found in 16% of ICP cases.[61][72] More recently, studies have demonstrated involvement of BSEP mutations in at least 5% of cases.[71] The V444A polymorphism of ABCB11 in particular may lead to ICP by causing a reduction in hepatic BSEP expression and consequently decreased bile salt export.[61] Other notable mutations identified in ICP patients include ones in the farnesoid X receptor (FXR), a nuclear receptor in hepatocytes which activates transcription of MDR3 and BSEP upon binding intracellular bile acids, thereby increasing canalicular bile efflux.[61][73][69]
Mechanism
Bile is secreted by the liver to aid in the digestion of fats. Bile formation begins in bile canaliculi that form between two adjacent surfaces of liver cells (hepatocytes) similar to the terminal branches of a tree. The canaliculi join each other to form larger and larger structures, sometimes referred to as the canals of Hering, which themselves join to form small bile ductules that have an epithelial surface. The ductules join to form bile ducts that eventually form either the right main hepatic duct that drains the right lobe of the liver, or the left main hepatic duct draining the left lobe of the liver. The two ducts join to form the common hepatic duct, which in turn joins the cystic duct from the gall bladder, to give the common bile duct. This duct then enters the duodenum at the ampulla of Vater. In cholestasis, bile accumulates in the hepatic parenchyma.[74]
One of the most common causes of extrahepatic, or obstructive cholestasis, is biliary obstruction. This is better known as choledocholithiasis where gallstones become stuck in the common bile duct.
Hepatocyte transporters implicated in drug-induced cholestasis
Mechanisms of drug-induced cholestasis
Drugs may induce cholestasis by interfering with 1) hepatic transporters, 2) bile canaliculi dynamics, and/or 3) cell structure and protein localization.[24][75] Hepatic transporters are essential for maintaining enterohepatic bile flow and bile acid homeostasis.[76] Therefore, their direct inhibition by certain drugs may lead to cholestasis. Relevant transporters implicated include BSEP, MDR3, MRP2-4, and NTCP.[24][66]
Cholestasis can result from competitive inhibition of BSEP by several drugs, including cyclosporine A, rifampicin, nefazodone, glibenclamide, troglitazone, and bosentan.[27][75] BSEP is the main transporter in hepatocytes responsible for exporting bile salts across the apical membrane into bile canaliculi. Therefore, inhibiting BSEP should cause cytotoxic bile salts to accumulate in hepatocytes, leading to liver injury and impaired bile flow.[27] Indeed, there is a strong association between BSEP inhibition and cholestasis in humans, and BSEP inhibitors are shown to induce cholestasis in vitro.[75] However, hepatocytes have safety mechanisms that can compensate for impaired canalicular bile efflux.[75] In response to cholestasis, MRP3 and MRP4 on the basolateral membrane are upregulated to allow efflux of accumulated bile salts into portal blood. Similarly, MRP2 can accommodate additional bile flow across the apical membrane in cholestatic conditions.[66][75] These compensatory mechanisms explain why some BSEP inhibitors do not cause cholestasis.[27][75] On the contrary, contrast, drugs that inhibit both MRP3/4 and BSEP (e.g., rifampicin, troglitazone, bosentan) pose greater risk for cholestasis[66]
MDR3 is another key canalicular efflux transporter that is the target of inhibition by certain drugs. MDR3 secretes phosphatidylcholine into bile canaliculi, where it form micelles with bile salts to dissolve cholesterol as well as protect hepatocyte and cholangiocytes from damage by bile salts.[24] MDR3 inhibition leads to low phospholipid concentrations in bile that damages cholangiocytes and leads to cholestasis.[77] Antifungal azoles such itraconazole have been shown to inhibit both MDR3 and BSEP, thus giving them higher cholestatic potential.[77] Other MDR3-inhibiting drugs are chlorpromazine, imipramine, haloperidol, ketoconazole, saquinavir, clotrimazole, ritonavir, and troglitazone.[27]
Another target for inhibition, MRP2 is an apical efflux transporter that mainly exports bilirubin glucuronide and glutathione into bile. However, MRP2 is also the preferential route of export for certain sulfated conjugated BAs (taurolithocholic acid and glycolithocholic acid), so its inhibition could contribute to cholestasis.[66]
On the hepatocyte basolateral membrane, Na+-taurocholate cotransporting peptide (NTCP) is the major transporter of conjugated bile acids.[75] Enterohepatic bile flow requires the concerted activity of both NTCP and BSEP, which form the major route by which BAs enter and exit hepatocytes respectively.[66] Therefore, NTCP inhibitors, such as cyclosporine A, ketoconazole, propranolol, furosemide, rifamycin, saquinavir, and ritonavir, should theoretically cause cholestasis by decreasing hepatocyte BA uptake.[75] However, no relationship was found between NTCP inhibition and DIC risk,[75] possibly because basolateral sodium-independent OATPs can partially compensate for bile salt uptake.[75] Therefore, NTCP inhibition alone seems to be insufficient for causeing cholestasis.[75] Indeed, the cholestatic effect of cyclosporine A relies on its inhibition of both NTCP and the compensatory OATP1B1.[75][77]
In addition to direct inhibition, drugs can also induce cholestasis by promoting downregulation and internalization of transporters. For example, cyclosporine A in rats was shown to induce BSEP internalization in addition to inhibition. Furthermore, human hepatocytes showed decreased expression of BSEP mRNA and protein following long-term exposure to metformin and tamoxifen, neither of which are direct BSEP inhibitors.[75]
Bile canaliculi dynamics refers to the contractile motion of bile canaliculi (ducts) required for bile flow. Cholestasis can result when drugs constrict or dilate bile canaliculi. Constrictors include chlorpromazine, nefazodone, troglitazone, perhexiline, metformin, cyclosporin A. These drugs activate the RhoA/Rho-kinase pathway, which inhibits myosin light chain phosphatase (MLCP), and in turn, increases myosin light chain phosphorylation by MLC kinase leading to constriction of bile canaliculi. Drugs that dilate canaliculi work by inhibiting MLCK or RhoA/Rho-kinase and include diclofenac, bosentan, entacapone, tacrolimus, cimetidine, and flucloxacillin.[66][24] Constriction is more serious than dilation, as the former causes irreversible cell damage and death.[24]
Minor mechanisms that may contribute to DIC include aberrant paracellular permeability, membrane fluidity, and transporter localization.[24]Tight junctions normally seal the gap between hepatocytes to prevent bile from diffusing out of the canaliculi. If a drug causes internalization of hepatocyte tight junctions, like rifampicin does in mice, bile flow may become impaired due to paracellular leakage.[66] Membrane fluidity can affect bile flow by regulating the activity of hepatocyte Na+/K+-ATPase, which maintains the inwardly-directed Na+ gradient that drives BA uptake by apical NTCP.[66][75] In rats, cyclosporine A was found to increase canalicular membrane fluidity and consequently reduce bile secretion. Bile flow was similarly reduced in rats as a result of alterations to basolateral membrane fluidity by ethinylestradiol and chlorpromazine.[66] Lastly, some agents (rimpaficin and 17β-estradiol) were shown to hinder proper localization of hepatocyte transporters by interfering with the microtubules required for their insertion into plasma membranes.[24]
Diagnosis
The diagnosis approach for identifying the cause of cholestasis
Cholestasis can be suspected when there is an elevation of both 5'-nucleotidase and ALP enzymes.[78] With a few exceptions, the optimal test for cholestasis would be elevations of serum bile acid levels.[79] However, this is not normally available in most clinical settings necessitating the use of other biomarkers. If 5' nucleosidase and ALP enzymes are elevated, imaging studies such as computed tomography (CT) scan, ultrasound, and magnetic resonance imaging (MRI) are used to differentiate intrahepatic cholestasis from extrahepatic cholestasis.[78] Additional imaging, laboratory testing, and biopsies might be conducted to identify the cause and extent of cholestasis.[78]
Biomarkers
ALP enzymes are found abundantly within the bile canaliculi and bile. If a duct is obstructed, tight junctions permit migration of the ALP enzymes until the polarity is reversed and the enzymes are found on the whole of the cell membrane.[78] Serum ALP levels exceeding 2–3 times the upper baseline value may be due to a variety of liver diseases.[80] However, an elevation that exceeds 10 times the upper baseline limit is strongly indicative of either intrahepatic or extrahepatic cholestasis and requires further investigation.[80] Cholestasis can be differentiated from other liver disorders by measuring the proportion of ALP to serum aminotransferases, where a greater proportion indicates a higher likelihood of cholestasis.[78] Typically, aminotransferase enzymes are localized within hepatocytes and leak across the membrane upon damage.[81] However, measurement of serum aminotransferase levels alone is not a good marker to determine cholestasis. In up to a third of patients, ALP levels may be elevated without the presence of cholestasis.[80] As such, other biomarkers should be measured to corroborate findings.
Measurement of 5' nucleosidase levels may be used to identify cholestasis in conjunction with ALP. Levels of ALP may rise within a few hours of cholestasis onset while 5' nucleosidase levels may take a few days.[82] Many labs cannot measure 5' nucleosidase and ALP levels so, GGT may be measured in some cases.[78] Abnormal GGT elevation may be attributable to a variety of factors.[83] As such, GGT elevations lack the necessary specificity to be a useful confirmatory test for cholestasis.[78]
Importantly, conjugated hyperbilirubinemia is present in 80% of patients with extrahepatic cholestasis and 50% of patients with intrahepatic cholestasis.[80] Given that many patients with hyperbilirubinemia may not have cholestasis, the measurement of bilirubin levels is not a good diagnostic tool for identifying cholestasis.[78] In a later stage of cholestasis aspartate transaminase (AST), alanine transaminase (ALT) and unconjugated bilirubin may be elevated due to hepatocyte damage as a secondary effect of cholestasis.
Imaging
After determination using biomarkers, a variety of imaging studies may be used to differentiate between intrahepatic or extrahepatic cholestasis. Ultrasound is often used to identify the location of the obstruction[84] but, is often insufficient in determining the level of biliary obstruction or its cause because it can pick up bowel gas that may interfere with readings.[78][85]CT scans are not impacted by bowel gas and may also be more suitable for overweight patients.[78] Typically, the cause of cholestasis and magnitude of obstruction is better diagnosed with CT compared to ultrasound.[86]MRI scans provide similar information to CT scans but are more prone to interference from breathing or other bodily functions.[87]
Although CT, ultrasound, and MRI may help differentiate intrahepatic and extrahepatic cholestasis, the cause and extent of obstruction is best determined by cholangiography.[78] Potential causes of extrahepatic cholestasis include obstructions outside the wall of the lumen, those outside the duct, and obstructions found in the duct lumen.[78] Endoscopic retrograde cholangiography may be useful to visualize the extrahepatic biliary ducts.[88] In case of anatomical anomalies, or if endoscopic retrograde cholangiography is unsuccessful, percutaneous transhepatic cholangiography may be used.[78] CT or MRI-based cholangiography may also be useful, particularly in cases where additional interventions are not anticipated.[78]
Histopathology
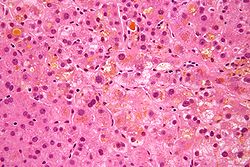
There is a significant overlap between cholestasis resulting from a hepatocellular origin and cholestasis caused by bile duct obstruction. Due to this, obstructive cholestasis can only be diagnosed after finding additional diagnostic signs that are specific to obstructive changes to the bile ducts or portal tracts.[89] In both non-obstructive and obstructive cholestasis, there is an accumulation of substances that are typically secreted in the bile, as well as degeneration of hepatocytes.[90] The most significant feature from a histopathological perspective includes pigmentation resulting from the retention of bilirubin. Under a microscope, the individual hepatocytes will have a brownish-green stippled appearance within the cytoplasm, representing bile that cannot get out of the cell. Pigmentation can involve regurgitation of bile into the sinusoidal spaces caused by phagocytosis from Kupffer cells, an accumulation of bilirubin within hepatocytes, and inspissated bile in the canaliculi.[89] Most pigmentation and canaliculi dilation occurs in the perivenular region of the hepatic lobule. In chronic cases, this may extend into the periportal area.[89]
Hepatocyte necrosis is not a significant feature of cholestasis; however, apoptosis may often occur.[91] Under the microscope, hepatocytes in the perivenular zone appear enlarged and flocculent.[89] In cases of obstructive cholestasis, bile infarcts may be produced during the degeneration and necrosis of hepatocytes.[91] Bile infarcts are marked by a large amount of pigmented tissue surrounded by a ring of necrotic hepatocytes.[89] In some cases, hepatocyte degeneration is uncommon. E.g., with Alagille syndrome limited degeneration occurs, however, there may be a small amount of apoptosis and enlarged hepatocytes.[92]
Cholestasis is often marked by cholate stasis, which are a set of changes that occur in the periportal hepatocytes.[93] Cholate stasis is more common in obstructive cholestasis compared to non-obstructive cholestasis.[94] During the cholate stasis process, hepatocytes first undergo swelling and then degeneration.[95] Under the microscope, this is evident as a lucent cell periphery and enlarged cytoplasm around the nucleus.[94] Oftentimes, Mallory bodies may also be found in the periportal areas.[96] Due to the retention of bile, which contains copper, stains made for staining copper-associated protein can be used to visualize bile accumulation in the hepatocytes.[97]
Cholestatic liver cell rosettes may occur in children with chronic cholestasis.[98] Histologically, this is evident as two or more hepatocytes in a pseudotubular fashion that encircle a segment of enlarged bile canaliculi.[94] Children may also have giant hepatocytes present, which are characterized by a pigmented spongy appearance.[99] Giant cell formation is likely caused by the detergent properties of bile salts causing a loss of the lateral membrane and joining of hepatocytes.[100] In the case of Alagille syndrome, hepatocyte degeneration is uncommon.[92] However, there may be a small amount of apoptosis and enlarged hepatocytes.[94]
In non-obstructive cholestasis, changes to the portal tracts are unlikely.[94] However, it may occur in some unique situations. In the case of neutrophilic pericholangitis, neutrophils surround the portal ducts and obstruct them.[94] Neutrophilic pericholangitis has a variety of causes including endotoxemia, Hodgkin's disease, among others.[101][102] Cholangitis lenta can also cause changes to the portal tracts.[103] This occurs during chronic cases of sepsis and results in dilation of the bile ductules.[94] Cholangitis lenta is likely a result of a stoppage of bile secretion and bile flow through the ductules.
Back pressure created from obstructive cholestasis can cause dilation of the bile duct and biliary epithelial cell proliferation, mainly in the portal tracts.[104] Portal tract edema may also occur as a result of bile retention, as well as periductular infiltration of neutrophils.[94] If the obstruction is left untreated, it can lead to a bacterial infection of the biliary tree.[105] Infection is mostly caused by coliforms and enterococci and is evident from a large migration of neutrophils to the duct lumina.[94] This can result in the formation of a cholangitic abscess. With treatment, many of the histological features of cholestasis can be corrected once the obstruction is removed.[94] If the obstruction is not promptly resolved, portal tract fibrosis can result. Even with treatment, some fibrosis may remain.[94]
Management
Surgical management
In cases involving obstructive cholestasis, the primary treatment includes biliary decompression.[106] If bile stones are present in the common bile duct, an endoscopic sphincterotomy can be conducted either with or without placing a stent.[2] To do this, a duodenoscope is placed by the endoscopist in the second portion of the duodenum. A catheter and guidewire is moved up into the common bile duct. A sphincterotome can then enlarge the ampulla of Vater and release the stones.[107] Later, the endoscopist can place a stent in the common bile duct to soften any remaining stones and allow for bile drainage. If needed, a balloon catheter is available to remove any leftover stones. If these stones are too large with these methods, surgical removal may be needed. Patients can also request an elective cholecystectomy to prevent future cases of choledocholithiasis.[108][109] In case of narrowing of the common bile duct, a stent can be placed after dilating the constriction to resolve the obstruction.[110]
The treatment approach for patients with obstructive cholestasis resulting from cancer varies based on whether they are a suitable candidate for surgery. In most cases, surgical intervention is the best option.[2] For patients whom complete removal of the biliary obstruction is not possible, a combination of a gastric bypass and hepaticojejunostomy can be used.[111] This can reestablish bile flow into the small intestine, thereby bypassing the blockage. In cases where a patient is not a suitable candidate for surgery, an endoscopic stent can be placed.[112] If this is not possible or successful, a percutaneous transhepatic cholangiogram and percutaneous biliary drainage can be used to visualize the blockage and re-establish bile flow.[113]
Medical management
A significant portion of patients with cholestasis (80%) will experience pruritus at some point during their disease.[114][115] This is a condition that can severely decrease a patient's quality of life as it can impact sleep, concentration, work ability, and mood. Many treatments exist, but how effective each option is depends on the patient and their condition. Assessment using a scale, such as a visual analogue scale or a 5-D itch scale will be useful to identify an appropriate treatment.[116] Possible treatment options include antihistamines, ursodeoxycholic acid, and phenobarbital. Nalfurafine hydrochloride can also be used to treat pruritus caused by chronic liver disease and was recently approved in Japan for this purpose.[117]
Bile acid binding resins like cholestyramine are the most common treatment. Side effects of this treatment are limited and include constipation and bloating. Other commonly used treatments include rifampin, naloxone, and sertraline.
In cholestatic liver disease, when bilirubin concentration starts to build up, a deficiency of fat soluble vitamins may also occur.[118] To manage this, doses of vitamin A, D, E, and K are recommended to retain appropriate vitamin levels.
Cholestatic liver disease can impact lipids, and possibly lead to dyslipidemia, which may present a risk for coronary artery disease.[119]Statins and fibrates are generally used as lipid lowering therapy to treat patients with cholestatic liver disease.
For intrahepatic cholestasis in pregnant women, S-adenosylmethionine has proven to be an effective treatment.[120]Dexamethasone is a viable treatment in regards to the symptom of intensive itching.[121]
Research directions
Primary sclerosing cholangitis (PSC) is one of the most common cholestatic liver diseases, yet treatment options remain limited. Treatment for primary biliary cholangitis (PBC) is often done with ursodeoxycholic acid (UDCA) and with no other suitable alternative, it poses a problem for those that are not responsive to (UDCA). However, with advancing technology in the molecular biochemistry field and higher understanding of bile acid regulation, novel pharmacological treatments have been considered.
For patients with primary biliary cholangitis, current guidelines recommend about 13–15mg/kg of ursodeoxycholic acid as a first line treatment.[122] This drug stimulates biliary bicarbonate secretion, improves survival without having to resort to a liver transplantation, and is very well tolerated—making it an ideal treatment.[123] However, around 40% of patients with primary biliary cholangitis are not responsive to UDCA.[124]
Obeticholic acid has been approved by the US Food and Drug Administration for PBC in 2016 after experiments found beneficial improvements for the liver in half of patients with inadequate response to UDCA.[125]
Primary sclerosing cholangitis is a challenging liver disease as treatment options are limited. There is still uncertainty about the efficacy of ursodeoxycholic acid for PSC and researchers offer conflicting recommendations.[126] One study found UDCA had improved biochemical functions but did lower the rate for death or transplant-free survival.[127]
Peroxisomes receptor agonists
An important regulator in bile acid homeostasis is the alpha and delta isoforms of the peroxisome proliferator-activated receptor (PPARα, PPARδ).[128] The function of PPARα is that it promotes bile acid excretion and lowers inflammation by acting on nuclear transcription factors.[129] A well known agonist are fibrates and in the clinical trials, there was a significant biochemical response in most patients.[130] A combination therapy with bezafibrate showed remarkable biochemical improvement, with 67% of patients normalizing their alkaline phosphatase levels.[131] Another study of 48 patients with PBC found a combination of bezafibrate and UDCA showed a decrease of alkaline phosphatase in all patients. Further, the study found those treated had a marked relief in pruritus.[132]
A new novel treatment option is the farnesoid X receptor is responsible for regulating bile acid homeostasis. An agonist of this nuclear hormone receptor is seen as a possible treatment as it can downregulate bile acid synthesis and reabsorption.[134] Further the farnesoid X receptor is partly responsible for lipid and glucose homeostasis, as well as pathogen recognition.[135] An agonist for the farnesoid X receptor can therefore lead to an anti-cholestatic environment to minimize the effect of toxic bile acids on the liver.[129] A candidate agonist for the farnesoid X receptor is obeticholic acid with experiments showing it has very strong affinity.[125] A worry though is despite benefits in the biochemical pathways, pruritus was more intense and prevalent than the placebo. A titration strategy may help to mitigate pruritus, but FDA approval for obeticholic acid is currently unlikely.[136] In fact, in February 2018, the FDA gave a black box warning for OCA.[137] A recent study did find that if the drug is given with UDCA, the incidence for cirrhosis and liver transplants decreases.[138]
Another target that is being looked into is the all-trans retinoic acid (ATRA), an activator for the retinoid X receptor. In vitro and animal studies found ATRA had lowered the amount of bile acid and decreased hepatic inflammation.[139]
24-norursodeoxycholic acid
A recent scientific breakthrough for cholestasis that has allowed us to evaluate a new treatment option is that a hydrophilic environment and bicarbonate production protects hepatocytes from bile acid.[128] The novel agent norUDCA (24-norursodeoxycholic acid) can be passively absorbed by cholangiocytes. This leads to bicarbonate production and an environment that is less toxic.[140][141] Mouse models have found promising results with norUDCA with the drug showing antiproliferative and anti-inflammatory properties. A recent clinical trial found norUDCA had significant dose-dependent reductions for ALP levels.[142] This makes norUDCA a viable possibility to look into as it clearly plays a significant role in the treatment of cholestasis.
Immunomodulatory treatments
In PBC, the liver is filled with T cells and B cells that contribute to a worsening condition.[143] Therefore, some treatments are looking into targeting the antigens of these immune cells. The monoclonal antibody rituximab targets the CD20 antigen on the B cells, and is already used in a wide array of other rheumatologic diseases. In an open-label study, six patients that were unresponsive to UDCA had improvement in ALP levels after rituximab infusions.[144] However, the efficacy of rituximab is still uncertain, and awaits further studies and trials. PBC can also lead to higher levels of interleukin 12 and interleukin 23. [145] This was what motivated researches to look at the viability of ustekinumab, a monoclonal antibody targeted against interleukin 12 and 23. An experiment found though it did not significantly improve serum ALP levels. The researchers were further even criticized for placing patients at risk by allowing them to move to advanced disease stages where immunomodulatory therapies may not even be an option.[146]
Gut microbiome
In several chronic liver diseases, the gut microbiome, which regulates both the innate and adaptive immune systems, is implicated. This can result in abnormal immunological development and an accumulation of primary bile acids. Using this information a bile-acid–intestinal-microbiota–cholestasis triangle is thought to be involved in the pathogenesis of PBC and PSC.[147] After all, bile acids do modulate the gut microbiota; a disturbance here can result in development and progression of cholestasis. This information has prompted researchers into manipulating the microbiota via antibiotics and probiotics for new treatment options. Some antibiotics examined for PSC include vancomycin, which has extensively studied and reviewed.[148] The usage of the drug is found along with a significant decrease in ALP levels, although the long term clinical benefit is unknown.[149]
As biochemistry technology becomes more advanced, promising targets have appeared, prompting numerous studies and trials to evaluate the feasibility. Fibrates, FXR agonists, and norUDCA are all innovative therapies for cholestasis.
↑ Brady P (May 2016). "Commentary on "Prospective Evaluation of the Clinical Features of Choledocholithiasis: Focus on Abdominal Pain"". Southern Medical Journal. 109 (5): 294–295. doi:10.14423/smj.0000000000000462. PMID27135725.
↑ Pearson HJ, Mosser JL, Jacks SK (November 2017). "The triad of pruritus, xanthomas, and cholestasis: Two cases and a brief review of the literature". Pediatric Dermatology. 34 (6): e305 –e308. doi:10.1111/pde.13306. PMID29144045. S2CID13254835.
↑ Sokol RJ (December 1994). "Fat-soluble vitamins and their importance in patients with cholestatic liver diseases". Gastroenterology Clinics of North America. 23 (4): 673–705. doi:10.1016/s0889-8553(21)00165-5. PMID7698827.
1 2 3 4 5 6 Fernández-Murga ML, Petrov PD, Conde I, Castell JV, Goméz-Lechón MJ, Jover R (October 2018). "Advances in drug-induced cholestasis: Clinical perspectives, potential mechanisms and in vitro systems". Food and Chemical Toxicology. 120: 196–212. doi:10.1016/j.fct.2018.07.017. PMID29990576. S2CID51612789.
1 2 "Cholestatic Hepatitis". LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases. 2012. PMID31689040. Retrieved 2021-11-16.
↑ "Vanishing Bile Duct Syndrome", LiverTox: Clinical and Research Information on Drug-Induced Liver Injury, Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases, 2012, PMID31689039, retrieved 2021-12-05
↑ "Bland Cholestasis", LiverTox: Clinical and Research Information on Drug-Induced Liver Injury, Bethesda (MD): National Institute of Diabetes and Digestive and Kidney Diseases, 2012, PMID31689032, retrieved 2021-11-16
↑ Jacquemin E (September 2012). "Progressive familial intrahepatic cholestasis". Clinics and Research in Hepatology and Gastroenterology. 36 (Suppl 1): S26 –S35. doi:10.1016/s2210-7401(12)70018-9. PMID23141890.
↑ Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E (May 2010). "The spectrum of liver diseases related to ABCB4 gene mutations: pathophysiology and clinical aspects". Seminars in Liver Disease. 30 (2): 134–146. doi:10.1055/s-0030-1253223. PMID20422496. S2CID260314414.
↑ Guglielmi FW, Regano N, Mazzuoli S, Fregnan S, Leogrande G, Guglielmi A, etal. (February 2008). "Cholestasis induced by total parenteral nutrition". Clinics in Liver Disease. 12 (1): 97–110, viii. doi:10.1016/j.cld.2007.11.004. PMID18242499.
↑ Cavicchi M, Beau P, Crenn P, Degott C, Messing B (April 2000). "Prevalence of liver disease and contributing factors in patients receiving home parenteral nutrition for permanent intestinal failure". Annals of Internal Medicine. 132 (7): 525–532. doi:10.7326/0003-4819-132-7-200004040-00003. PMID10744588. S2CID26657365.
↑ Jeejeebhoy KN (February 2006). "Management of short bowel syndrome: avoidance of total parenteral nutrition". Gastroenterology. 130 (2 Suppl 1): S60 –S66. doi:10.1053/j.gastro.2005.10.065. PMID16473074.
1 2 3 Dixon PH, Williamson C (April 2016). "The pathophysiology of intrahepatic cholestasis of pregnancy". Clinics and Research in Hepatology and Gastroenterology. 40 (2): 141–153. doi:10.1016/j.clinre.2015.12.008. PMID26823041.
1 2 Alagille D, Odièvre M, Gautier M, Dommergues JP (January 1975). "Hepatic ductular hypoplasia associated with characteristic facies, vertebral malformations, retarded physical, mental, and sexual development, and cardiac murmur". The Journal of Pediatrics. 86 (1): 63–71. doi:10.1016/s0022-3476(75)80706-2. PMID803282.
↑ Lefkowitch JH (January 1982). "Bile ductular cholestasis: an ominous histopathologic sign related to sepsis and "cholangitis lenta"". Human Pathology. 13 (1): 19–24. doi:10.1016/s0046-8177(82)80134-2. PMID7076191.
↑ Parikh MP, Gupta NM, Thota PN, Lopez R, Sanaka MR (April 2018). "Temporal trends in utilization and outcomes of endoscopic retrograde cholangiopancreatography in acute cholangitis due to choledocholithiasis from 1998 to 2012". Surgical Endoscopy. 32 (4): 1740–1748. doi:10.1007/s00464-017-5856-7. PMID28917018. S2CID39106004.
↑ Benites Goñi HE, Palacios Salas FV, Asencios Cusihuallpa JL, Aguilar Morocco R, Segovia Valle NS (April 2017). "[Performance of ASGE predictive criteria in diagnosis of choledocholithiasis in the Edgardo Rebagliati Martins Hospital]". Revista de Gastroenterologia del Peru. 37 (2): 111–119. PMID28731990.
↑ Voegeli DR, Crummy AB, Weese JL (August 1985). "Percutaneous transhepatic cholangiography, drainage, and biopsy in patients with malignant biliary obstruction. An alternative to surgery". American Journal of Surgery. 150 (2): 243–247. doi:10.1016/0002-9610(85)90129-1. PMID2411158.
↑ Hirvioja ML, Tuimala R, Vuori J (February 1992). "The treatment of intrahepatic cholestasis of pregnancy by dexamethasone". British Journal of Obstetrics and Gynaecology. 99 (2): 109–111. doi:10.1111/j.1471-0528.1992.tb14465.x. PMID1554659. S2CID2391542.
↑ Floreani A, Mangini C (January 2018). "Primary biliary cholangitis: Old and novel therapy". European Journal of Internal Medicine. 47: 1–5. doi:10.1016/j.ejim.2017.06.020. PMID28669591.
↑ Corpechot C, Abenavoli L, Rabahi N, Chrétien Y, Andréani T, Johanet C, etal. (September 2008). "Biochemical response to ursodeoxycholic acid and long-term prognosis in primary biliary cirrhosis". Hepatology. 48 (3): 871–877. doi:10.1002/hep.22428. PMID18752324. S2CID40564903.
↑ Reig A, Sesé P, Parés A (January 2018). "Effects of Bezafibrate on Outcome and Pruritus in Primary Biliary Cholangitis With Suboptimal Ursodeoxycholic Acid Response". The American Journal of Gastroenterology. 113 (1): 49–55. doi:10.1038/ajg.2017.287. PMID29016567. S2CID205105216.
↑ Iwasaki S, Ohira H, Nishiguchi S, Zeniya M, Kaneko S, Onji M, etal. (June 2008). "The efficacy of ursodeoxycholic acid and bezafibrate combination therapy for primary biliary cirrhosis: A prospective, multicenter study". Hepatology Research. 38 (6): 557–564. doi:10.1111/j.1872-034X.2007.00305.x. PMID18452482. S2CID36861916.
↑ Trauner M, Fickert P, Halilbasic E, Moustafa T (2008). "Lessons from the toxic bile concept for the pathogenesis and treatment of cholestatic liver diseases". Wiener Medizinische Wochenschrift. 158 (19–20): 542–548. doi:10.1007/s10354-008-0592-1. PMID18998069. S2CID25907527.
↑ Dumont M, Erlinger S, Uchman S (July 1980). "Hypercholeresis induced by ursodeoxycholic acid and 7-ketolithocholic acid in the rat: possible role of bicarbonate transport". Gastroenterology. 79 (1): 82–89. doi:10.1016/0016-5085(80)90078-5. PMID7380227.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.