The mGluRs perform a variety of functions in the central and peripheral nervous systems: For example, they are involved in learning, memory, anxiety, and the perception of pain.[2] They are found in pre- and postsynaptic neurons in synapses of the hippocampus, cerebellum,[3] and the cerebral cortex, as well as other parts of the brain and in peripheral tissues.[4]
Eight different types of mGluRs, labeled mGluR1 to mGluR8 (GRM1 to GRM8), are divided into groups I, II, and III.[1][3][4][8] Receptor types are grouped based on receptor structure and physiological activity.[2] The mGluRs are further divided into subtypes, such as mGluR7a and mGluR7b.
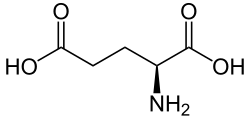
Group I mGluRs, but not other groups, are activated by 3,5-dihydroxyphenylglycine (DHPG),[14] a fact that is useful to experimenters because it allows them to isolate and identify them.
Group II and Group III
The receptors in group II, including mGluRs 2 and 3, and group III, including mGluRs 4, 6, 7, and 8, (with some exceptions) prevent the formation of cyclic adenosine monophosphate, or cAMP, by activating a G protein that inhibits the enzyme adenylyl cyclase, which forms cAMP from ATP.[1][3][4][17] These receptors are involved in presynaptic inhibition,[8] and do not appear to affect postsynaptic membrane potential by themselves. Receptors in groups II and III reduce the activity of postsynaptic potentials, both excitatory and inhibitory, in the cortex.[4]
LY-341,495 and MGS-0039 are drugs that act as a selective antagonist blocking both of the group II metabotropic glutamate receptors, mGluR2 and mGluR3.[18]RO4491533 acts as a negative allosteric modulator of mGluR2 and mGluR3.[19]
Localization
Different types of mGluRs are distributed differently in cells. For example, one study found that Group I mGluRs are located mostly on postsynaptic parts of cells, while groups II and III are mostly located on presynaptic elements,[14] though they have been found on both pre- and postsynaptic membranes.[8]
Also, different mGluR subtypes are found predominantly in different parts of the body. For example, mGluR4 is located only in the brain, in locations such as the thalamus, hypothalamus and caudate nucleus.[20] All mGluRs except mGluR6 are thought to exist in the hippocampus and entorhinal cortex.[14]
Roles
It is thought that mGluRs play a role in a variety of different functions.
Modulation of other receptors
Metabotropic glutamate receptors are known to act as modulators of (affect the activity of) other receptors. For example, group I mGluRs are known to increase the activity of N-methyl-D-aspartate receptors (NMDARs),[12][13] a type of ion channel-linked receptor that is central in a neurotoxic process called excitotoxicity. Proteins called PDZ proteins frequently anchor mGluRs near enough to NMDARs to modulate their activity.[21]
It has been suggested that mGluRs may act as regulators of neurons' vulnerability to excitotoxicity (a deadly neurochemical process involving glutamate receptor overactivation) through their modulation of NMDARs, the receptor most involved in that process.[22] Excessive amounts of N-methyl-D-aspartate (NMDA), the selective specific agonist of NMDARs, has been found to cause more damage to neurons in the presence of group I mGluR agonists.[23] On the other hand, agonists of group II[24] and III mGluRs reduce NMDAR activity.[15]
Group II[25] and III[23] mGluRs tend to protect neurons from excitotoxicity,[15][26][27] possibly by reducing the activity of NMDARs.
Metabotropic glutamate receptors are also thought to affect dopaminergic and adrenergic neurotransmission.[28]
Since metabotropic glutamate receptors are involved in a variety of functions, abnormalities in their expression can contribute to disease. For example, studies with mutant mice have suggested that mutations in expression of mGluR1 may be involved in the development of certain types of cancer.[31] In addition, manipulating mGluRs can be useful in treating some conditions. For example, clinical trial suggested that an mGlu2/3 agonist, LY354740, was effective in the treatment of generalized anxiety disorder.[32] Also, some researchers have suggested that activation of mGluR4 could be used as a treatment for Parkinson's disease.[33] Most recently, Group I mGluRs, have been implicated in the pathogenesis of Fragile X, a type of autism,[34] and a number of studies are currently testing the therapeutic potential of drugs that modify these receptors.[35] There is also growing evidence that group II metabotropic glutamate receptor agonists may play a role in the treatment of schizophrenia. Schizophrenia is associated with deficits in cortical inhibitory interneurons that release GABA and synaptic abnormalities associated with deficits in NMDA receptor function.[36] These inhibitory deficits may impair cortical function via cortical disinhibition and asynchrony.[37] The drug LY354740 (also known as Eglumegad, an mGlu2/3agonist) was shown to attenuate physiologic and cognitive abnormalities in animal and human studies of NMDA receptor antagonist and serotonergic hallucinogen effects,[38][39][40][41] supporting the subsequent clinical evidence of efficacy for an mGluR2/3 agonist in the treatment of schizophrenia.[42] The same drug has been shown to interfere in the hypothalamic–pituitary–adrenal axis, with chronic oral administration of this drug leading to markedly reduced baseline cortisol levels in bonnet macaques (Macaca radiata); acute infusion of LY354740 resulted in a marked diminution of yohimbine-induced stress response in those animals.[43]LY354740 has also been demonstrated to act on the metabotropic glutamate receptor 3 (GRM3) of human adrenocortical cells, downregulating aldosterone synthase, CYP11B1, and the production of adrenalsteroids (i.e. aldosterone and cortisol).[44]
History
The first demonstration that glutamate could induce the formation of molecules belonging to a major second messenger system was in 1985, when it was shown that it could stimulate the formation of inositol phosphates.[45] This finding allowed in 1987 to yield an explanation for oscillatory ionic glutamate responses and to provide further evidence for the existence of metabotropic glutamate receptors.[46] In 1991 the first metabotropic glutamate receptor of the seven transmembrane domain family was cloned.[47] More recent reports on ionotropic glutamate receptors able to couple to metabotropic transduction systems[48][49] suggest that metabotropic responses of glutamate might not be limited to seven transmembrane domain metabotropic glutamate receptors.
References
1 2 3 4 5 6 Bonsi P, Cuomo D, De Persis C, Centonze D, Bernardi G, Calabresi P, Pisani A (2005). "Modulatory action of metabotropic glutamate receptor (mGluR) 5 on mGluR1 function in striatal cholinergic interneurons". Neuropharmacology. 49. 49 (Suppl 1): 104–13. doi:10.1016/j.neuropharm.2005.05.012. PMID16005029. S2CID25980146.
1 2 3 Hinoi E, Ogita K, Takeuchi Y, Ohashi H, Maruyama T, Yoneda Y (March 2001). "Characterization with [3H]quisqualate of group I metabotropic glutamate receptor subtype in rat central and peripheral excitable tissues". Neurochemistry International. 38 (3): 277–85. doi:10.1016/S0197-0186(00)00075-9. PMID11099787. S2CID19402633.
1 2 Platt SR (March 2007). "The role of glutamate in central nervous system health and disease--a review". Veterinary Journal. 173 (2): 278–86. doi:10.1016/j.tvjl.2005.11.007. PMID16376594.
↑ Sladeczek F., Momiyama A.,Takahashi T. (1992). "Presynaptic inhibitory action of metabotropic glutamate receptor agonist on excitatory transmission in visual cortical neurons". Proc. Roy. Soc. Lond. B 1993 253, 297-303.
↑ Swanson CJ, Bures M, Johnson MP, Linden AM, Monn JA, Schoepp DD (February 2005). "Metabotropic glutamate receptors as novel targets for anxiety and stress disorders". Nature Reviews. Drug Discovery. 4 (2): 131–44. doi:10.1038/nrd1630. PMID15665858. S2CID15037387.
1 2 Skeberdis VA, Lan J, Opitz T, Zheng X, Bennett MV, Zukin RS (June 2001). "mGluR1-mediated potentiation of NMDA receptors involves a rise in intracellular calcium and activation of protein kinase C". Neuropharmacology. 40 (7): 856–65. doi:10.1016/S0028-3908(01)00005-3. PMID11378156. S2CID19635322.
1 2 Lea PM, Custer SJ, Vicini S, Faden AI (September 2002). "Neuronal and glial mGluR5 modulation prevents stretch-induced enhancement of NMDA receptor current". Pharmacology Biochemistry and Behavior. 73 (2): 287–98. doi:10.1016/S0091-3057(02)00825-0. PMID12117582. S2CID20086782.
↑ Koike H, Iijima M, Chaki S (December 2011). "Involvement of the mammalian target of rapamycin signaling in the antidepressant-like effect of group II metabotropic glutamate receptor antagonists". Neuropharmacology. 61 (8): 1419–23. doi:10.1016/j.neuropharm.2011.08.034. PMID21903115. S2CID23923192.
↑ Campo B, Kalinichev M, Lambeng N, El Yacoubi M, Royer-Urios I, Schneider M, Legrand C, Parron D, Girard F, Bessif A, Poli S, Vaugeois JM, Le Poul E, Celanire S (December 2011). "Characterization of an mGluR2/3 negative allosteric modulator in rodent models of depression". Journal of Neurogenetics. 25 (4): 152–66. doi:10.3109/01677063.2011.627485. PMID22091727. S2CID207440972.
↑ Baskys A, Blaabjerg M (March 2005). "Understanding regulation of nerve cell death by mGluRs as a method for development of successful neuroprotective strategies". Journal of the Neurological Sciences. 229–230: 201–9. doi:10.1016/j.jns.2004.11.028. PMID15760640. S2CID39812016.
1 2 Bruno V, Copani A, Knöpfel T, Kuhn R, Casabona G, Dell'Albani P, Condorelli DF, Nicoletti F (August 1995). "Activation of metabotropic glutamate receptors coupled to inositol phospholipid hydrolysis amplifies NMDA-induced neuronal degeneration in cultured cortical cells". Neuropharmacology. 34 (8): 1089–98. doi:10.1016/0028-3908(95)00077-J. PMID8532158. S2CID23848439.
↑ Buisson A, Yu SP, Choi DW (January 1996). "DCG-IV selectively attenuates rapidly triggered NMDA-induced neurotoxicity in cortical neurons". The European Journal of Neuroscience. 8 (1): 138–43. doi:10.1111/j.1460-9568.1996.tb01174.x. PMID8713457. S2CID24122616.
↑ Bruno V, Battaglia G, Copani A, Giffard RG, Raciti G, Raffaele R, Shinozaki H, Nicoletti F (September 1995). "Activation of class II or III metabotropic glutamate receptors protects cultured cortical neurons against excitotoxic degeneration". The European Journal of Neuroscience. 7 (9): 1906–13. doi:10.1111/j.1460-9568.1995.tb00712.x. PMID8528465. S2CID10181885.
↑ Faden AI, Ivanova SA, Yakovlev AG, Mukhin AG (December 1997). "Neuroprotective effects of group III mGluR in traumatic neuronal injury". Journal of Neurotrauma. 14 (12): 885–95. doi:10.1089/neu.1997.14.885. PMID9475370.
↑ Wang JQ, Brownell AL (2007). "Development of metabotropic glutamate receptor ligands for neuroimaging". Current Medical Imaging Reviews. 3 (3): 186–205. doi:10.2174/157340507781387059.
↑ Siliprandi R, Lipartiti M, Fadda E, Sautter J, Manev H (August 1992). "Activation of the glutamate metabotropic receptor protects retina against N-methyl-D-aspartate toxicity". European Journal of Pharmacology. 219 (1): 173–4. doi:10.1016/0014-2999(92)90598-X. PMID1397046.
↑ Baskys A, Fang L, Bayazitov I (August 2005). "Activation of neuroprotective pathways by metabotropic group I glutamate receptors: a potential target for drug discovery?". Annals of the New York Academy of Sciences. 1053 (1): 55–73. Bibcode:2005NYASA1053...55B. doi:10.1111/j.1749-6632.2005.tb00011.x. PMID16179509.
↑ Homayoun H, Jackson ME, Moghaddam B (April 2005). "Activation of metabotropic glutamate 2/3 receptors reverses the effects of NMDA receptor hypofunction on prefrontal cortex unit activity in awake rats". Journal of Neurophysiology. 93 (4): 1989–2001. doi:10.1152/jn.00875.2004. PMID15590730. S2CID5472650.
↑ Krystal JH, Abi-Saab W, Perry E, D'Souza DC, Liu N, Gueorguieva R, McDougall L, Hunsberger T, Belger A, Levine L, Breier A (April 2005). "Preliminary evidence of attenuation of the disruptive effects of the NMDA glutamate receptor antagonist, ketamine, on working memory by pretreatment with the group II metabotropic glutamate receptor agonist, LY354740, in healthy human subjects". Psychopharmacology. 179 (1): 303–9. doi:10.1007/s00213-004-1982-8. PMID15309376. S2CID29731346.
↑ Aghajanian GK, Marek GJ (March 2000). "Serotonin model of schizophrenia: emerging role of glutamate mechanisms". Brain Research. Brain Research Reviews. 31 (2–3): 302–12. doi:10.1016/S0165-0173(99)00046-6. PMID10719157. S2CID13040014.
↑ Patil ST, Zhang L, Martenyi F, Lowe SL, Jackson KA, Andreev BV, Avedisova AS, Bardenstein LM, Gurovich IY, Morozova MA, Mosolov SN, Neznanov NG, Reznik AM, Smulevich AB, Tochilov VA, Johnson BG, Monn JA, Schoepp DD (September 2007). "Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial". Nature Medicine. 13 (9): 1102–7. doi:10.1038/nm1632. PMID17767166. S2CID6417333.
↑ Coplan JD, Mathew SJ, Smith EL, Trost RC, Scharf BA, Martinez J, Gorman JM, Monn JA, Schoepp DD, Rosenblum LA (July 2001). "Effects of LY354740, a novel glutamatergic metabotropic agonist, on nonhuman primate hypothalamic-pituitary-adrenal axis and noradrenergic function". CNS Spectrums. 6 (7): 607–12, 617. doi:10.1017/S1092852900002157. PMID15573025. S2CID6029856.
↑ Felizola SJ, Nakamura Y, Satoh F, Morimoto R, Kikuchi K, Nakamura T, Hozawa A, Wang L, Onodera Y, Ise K, McNamara KM, Midorikawa S, Suzuki S, Sasano H (January 2014). "Glutamate receptors and the regulation of steroidogenesis in the human adrenal gland: the metabotropic pathway". Molecular and Cellular Endocrinology. 382 (1): 170–7. doi:10.1016/j.mce.2013.09.025. PMID24080311. S2CID3357749.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.