Related Research Articles

Lysosomal storage diseases are a group of over 70 rare inherited metabolic disorders that result from defects in lysosomal function. Lysosomes are sacs of enzymes within cells that digest large molecules and pass the fragments on to other parts of the cell for recycling. This process requires several critical enzymes. If one of these enzymes is defective due to a mutation, the large molecules accumulate within the cell, eventually killing it.

Adenosine deaminase deficiency is a metabolic disorder that causes immunodeficiency. It is caused by mutations in the ADA gene. It accounts for about 10–20% of all cases of autosomal recessive forms of severe combined immunodeficiency (SCID) after excluding disorders related to inbreeding.

Alpha-mannosidosis is a lysosomal storage disorder, first described by Swedish physician Okerman in 1967. In humans it is known to be caused by an autosomal recessive genetic mutation in the gene MAN2B1, located on chromosome 19, affecting the production of the enzyme alpha-D-mannosidase, resulting in its deficiency. Consequently, if both parents are carriers, there will be a 25% chance with each pregnancy that the defective gene from both parents will be inherited, and the child will develop the disease. There is a two in three chance that unaffected siblings will be carriers. In livestock alpha-mannosidosis is caused by chronic poisoning with swainsonine from locoweed.

Glycogen storage disease type II(GSD-II), also called Pompe disease, and formerly known as GSD-IIa or Limb–girdle muscular dystrophy 2V, is an autosomal recessive metabolic disorder which damages muscle and nerve cells throughout the body. It is caused by an accumulation of glycogen in the lysosome due to deficiency of the lysosomal acid alpha-glucosidase enzyme (GAA). The inability to break down glycogen within the lysosomes of cells leads to progressive muscle weakness throughout the body and affects various body tissues, particularly in the heart, skeletal muscles, liver and the nervous system.

Pyruvate kinase deficiency is an inherited metabolic disorder of the enzyme pyruvate kinase which affects the survival of red blood cells. Both autosomal dominant and recessive inheritance have been observed with the disorder; classically, and more commonly, the inheritance is autosomal recessive. Pyruvate kinase deficiency is the second most common cause of enzyme-deficient hemolytic anemia, following G6PD deficiency.
Inborn errors of metabolism form a large class of genetic diseases involving congenital disorders of enzyme activities. The majority are due to defects of single genes that code for enzymes that facilitate conversion of various substances (substrates) into others (products). In most of the disorders, problems arise due to accumulation of substances which are toxic or interfere with normal function, or due to the effects of reduced ability to synthesize essential compounds. Inborn errors of metabolism are often referred to as congenital metabolic diseases or inherited metabolic disorders. Another term used to describe these disorders is "enzymopathies". This term was created following the study of biodynamic enzymology, a science based on the study of the enzymes and their products. Finally, inborn errors of metabolism were studied for the first time by British physician Archibald Garrod (1857–1936), in 1908. He is known for work that prefigured the "one gene–one enzyme" hypothesis, based on his studies on the nature and inheritance of alkaptonuria. His seminal text, Inborn Errors of Metabolism, was published in 1923.

Adenosine deaminase is an enzyme involved in purine metabolism. It is needed for the breakdown of adenosine from food and for the turnover of nucleic acids in tissues.
Metachromatic leukodystrophy (MLD) is a lysosomal storage disease which is commonly listed in the family of leukodystrophies as well as among the sphingolipidoses as it affects the metabolism of sphingolipids. Leukodystrophies affect the growth and/or development of myelin, the fatty covering which acts as an insulator around nerve fibers throughout the central and peripheral nervous systems. MLD involves cerebroside sulfate accumulation. Metachromatic leukodystrophy, like most enzyme deficiencies, has an autosomal recessive inheritance pattern.
A lipid storage disorder is any one of a group of inherited metabolic disorders in which harmful amounts of fats or lipids accumulate in some body cells and tissues. People with these disorders either do not produce enough of one of the enzymes needed to metabolize and break down lipids or, they produce enzymes that do not work properly. Over time, the buildup of fats may cause permanent cellular and tissue damage, particularly in the brain, peripheral nervous system, liver, spleen, and bone marrow.

Roscoe Owen Brady was an American biochemist.

Hunter syndrome, or mucopolysaccharidosis type II, is a rare genetic disorder in which large sugar molecules called glycosaminoglycans build up in body tissues. It is a form of lysosomal storage disease. Hunter syndrome is caused by a deficiency of the lysosomal enzyme iduronate-2-sulfatase (I2S). The lack of this enzyme causes heparan sulfate and dermatan sulfate to accumulate in all body tissues. Hunter syndrome is the only MPS syndrome to exhibit X-linked recessive inheritance.

Deoxyadenosine triphosphate (dATP) is a nucleotide used in cells for DNA synthesis, as a substrate of DNA polymerase.
Purine nucleoside phosphorylase deficiency is a rare autosomal recessive metabolic disorder which results in immunodeficiency.
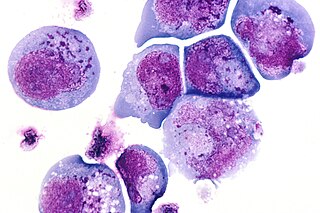
I-cells, also called inclusion cells, are abnormal fibroblasts having a large number of dark inclusions in the cytoplasm of the cell. Inclusion bodies are nuclear or cytoplasmic aggregates of stainable substances, usually proteins. These metabolically inactive aggregates are not enclosed by a membrane, and are composed of fats, proteins, carbohydrates, pigments, and excretory products. When cells have an abundance of these inclusions, they are called I-Cells and are associated with neurodegenerative diseases. They are seen in Mucolipidosis II, and Mucolipidosis III, also called inclusion-cell or I-cell disease where lysosomal enzyme transport and storage is affected.
Substrate reduction therapy offers an approach to treatment of certain metabolic disorders, especially glycogen storage diseases and lysosomal storage disorders. In a storage disorder, a critical failure in a metabolic pathway prevents cellular breakdown and disposal of some large molecule. If residual breakdown through other pathways is insufficient to prevent harmful accumulation, the molecule accumulates in the cell and eventually interferes with normal biological processes. Examples of lysosomal storage disorders include Gaucher's disease, Tay–Sachs disease, Sandhoff disease, and Sanfilippo syndrome.

Robert J. Desnick is an American human geneticist whose basic and translational research accomplishments include significant discoveries in genomics, pharmacogenetics, gene therapy, personalized medicine, and the treatment of genetic diseases. His translational research has led to the development of the enzyme replacement therapy (ERT) and the chaperone therapy for Fabry disease, ERT for Niemann–Pick disease type B, and the RNA Interference Therapy for the Acute Hepatic Porphyrias.
Lysosomal acid lipase deficiency or Wolman Disease, is an autosomal recessive inborn error of metabolism that results in the body not producing enough active lysosomal acid lipase (LAL) enzyme. This enzyme plays an important role in breaking down fatty material in the body. Infants, children and adults that have LAL deficiency experience a range of serious health problems. The lack of the LAL enzyme can lead to a build-up of fatty material in a number of body organs including the liver, spleen, gut, in the wall of blood vessels and other important organs.
Autologous CD34+ enriched cell fraction that contains CD34+ cells transduced with retroviral vector that encodes for the human ADA cDNA sequence, sold under the brand name Strimvelis, is a medication used to treat severe combined immunodeficiency due to adenosine deaminase deficiency (ADA-SCID).
Deficiency of Adenosine deaminase 2 (DADA2) is a monogenic disease associated with systemic inflammation and vasculopathy that affects a wide variety of organs in different patients. As a result, it is hard to characterize a patient with this disorder. Manifestations of the disease include but are not limited to recurrent fever, livedoid rash, various cytopenias, stroke, immunodeficiency, and bone marrow failure. Symptoms often onset during early childhood, but some cases have been discovered as late as 65 years old.
Olipudase alfa, sold under the brand name Xenpozyme, is a medication used for the treatment of non-central nervous system (CNS) manifestations of acid sphingomyelinase deficiency type A/B or type B.
References
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 Ries, M (2017). "Enzyme replacement therapy and beyond-in memoriam Roscoe O. Brady, M.D. (1923–2016)". Journal of Inherited Metabolic Disease. 40 (3): 343–356. doi:10.1007/s10545-017-0032-8. PMID 28314976. S2CID 31320877.
- 1 2 Booth, C; Hershfield, M; Notarangelo, L; Buckley, R; Hoenig, M; Mahlaoui, N; Cavazzana-Calvo, M; Aiuti, A; Gaspar, H. B. (2007). "Management options for adenosine deaminase deficiency; proceedings of the EBMT satellite workshop (Hamburg, March 2006)". Clinical Immunology. 123 (2): 139–47. doi:10.1016/j.clim.2006.12.009. PMID 17300989.
- ↑ Ries, M (2017). "Enzyme replacement therapy and beyond-in memoriam Roscoe O. Brady, M.D. (1923–2016)". Journal of Inherited Metabolic Disease. 40 (3): 343–356. doi:10.1007/s10545-017-0032-8. PMID 28314976. S2CID 31320877.[ verification needed ]
- ↑ Ries, M (2017). "Enzyme replacement therapy and beyond-in memoriam Roscoe O. Brady, M.D. (1923–2016)". Journal of Inherited Metabolic Disease. 40 (3): 343–356. doi:10.1007/s10545-017-0032-8. PMID 28314976. S2CID 31320877.[ verification needed ]
- ↑ Neufeld EF (2006). "Chapter 10: Enzyme replacement therapy – a brief history". In Mehta A, Beck M, Sunder-Plassmann G (eds.). Fabry Disease: Perspectives from 5 Years of FOS. Oxford: Oxford PharmaGenesis. ISBN 978-1-903539-03-3. PMID 21290685.
- ↑ Poznansky, M. J. (1984). "Enzyme-albumin polymers. New approaches to the use of enzymes in medicine". Applied Biochemistry and Biotechnology. 10: 41–56. doi:10.1007/BF02783734. PMID 6395807. S2CID 87349537.
- 1 2 3 4 Weiss, N (2012). "Cross-talk between TRPML1 channel, lipids and lysosomal storage diseases". Communicative & Integrative Biology. 5 (2): 111–113. doi:10.4161/cib.20373. PMC 3376041 . PMID 22808310.
- 1 2 "Lysosomal Storage Disorders – NORD (National Organization For Rare Disorders)". NORD (National Organization for Rare Disorders). Apr. 2017, from https://rarediseases.org/rare-diseases/lysosomal-storage-disorders/.
- 1 2 3 4 5 Tartibi, H. M.; Hershfield, M. S.; Bahna, S. L. (2016). "A 24-Year Enzyme Replacement Therapy in an Adenosine-deaminase-Deficient Patient". Pediatrics. 137 (1): e20152169. doi: 10.1542/peds.2015-2169 . PMID 26684479.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 Coutinho, M. F.; Santos, J. I.; Alves, S (2016). "Less is More: Substrate Reduction Therapy for Lysosomal Storage Disorders". International Journal of Molecular Sciences. 17 (7): 1065. doi: 10.3390/ijms17071065 . PMC 4964441 . PMID 27384562.
- 1 2 3 4 "How does gene therapy work? - Genetics Home Reference." U.S. National Library of Medicine. April 18, 2017, from https://ghr.nlm.nih.gov/primer/therapy/procedures.
- 1 2 3 Biffi, A (2016). "Gene therapy for lysosomal storage disorders: A good start". Human Molecular Genetics. 25 (R1): R65–75. doi: 10.1093/hmg/ddv457 . PMID 26604151.
- 1 2 Domen, J., Wagers, A., & Weissman, I. "Bone Marrow (Hematopoietic) Stem Cells." April 19, 2017, from https://stemcells.nih.gov/info/Regenerative_Medicine/2006chapter2.htm Archived 2021-05-15 at the Wayback Machine .