Related Research Articles
An exon is any part of a gene that will encode a part of the final mature RNA produced by that gene after introns have been removed by RNA splicing. The term exon refers to both the DNA sequence within a gene and to the corresponding sequence in RNA transcripts. In RNA splicing, introns are removed and exons are covalently joined to one another as part of generating the mature messenger RNA. Just as the entire set of genes for a species constitutes the genome, the entire set of exons constitutes the exome.

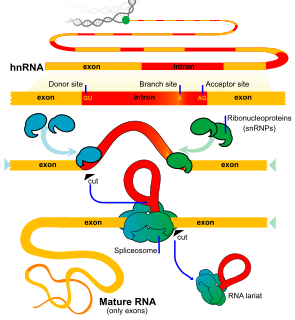
RNA splicing, in molecular biology, is a form of RNA processing in which a newly made precursor messenger RNA (pre-mRNA) transcript is transformed into a mature messenger RNA (mRNA). During splicing, introns are removed and exons are joined together. For nuclear-encoded genes, splicing takes place within the nucleus either during or immediately after transcription. For those eukaryotic genes that contain introns, splicing is usually required in order to create an mRNA molecule that can be translated into protein. For many eukaryotic introns, splicing is carried out in a series of reactions which are catalyzed by the spliceosome, a complex of small nuclear ribonucleoproteins (snRNPs). Self-splicing introns, or ribozymes capable of catalyzing their own excision from their parent RNA molecule, also exist.

The human genome is a complete set of nucleic acid sequences for humans, encoded as DNA within the 23 chromosome pairs in cell nuclei and in a small DNA molecule found within individual mitochondria. These are usually treated separately as the nuclear genome, and the mitochondrial genome. Human genomes include both protein-coding DNA genes and noncoding DNA. Haploid human genomes, which are contained in germ cells consist of three billion DNA base pairs, while diploid genomes have twice the DNA content. While there are significant differences among the genomes of human individuals, these are considerably smaller than the differences between humans and their closest living relatives, the bonobos and chimpanzees.
Non-coding DNA sequences are components of an organism's DNA that do not encode protein sequences. Some non-coding DNA is transcribed into functional non-coding RNA molecules. Other functions of non-coding DNA include the transcriptional and translational regulation of protein-coding sequences, scaffold attachment regions, origins of DNA replication, centromeres and telomeres. Its RNA counterpart is non-coding RNA.

Gene expression is the process by which information from a gene is used in the synthesis of a functional gene product. These products are often proteins, but in non-protein-coding genes such as transfer RNA (tRNA) or small nuclear RNA (snRNA) genes, the product is a functional RNA. Gene expression is summarized in the Central Dogma first formulated by Francis Crick in 1958, further developed in his 1970 article, and expanded by the subsequent discoveries of reverse transcription and RNA replication.

A non-coding RNA (ncRNA) is an RNA molecule that is not translated into a protein. The DNA sequence from which a functional non-coding RNA is transcribed is often called an RNA gene. Abundant and functionally important types of non-coding RNAs include transfer RNAs (tRNAs) and ribosomal RNAs (rRNAs), as well as small RNAs such as microRNAs, siRNAs, piRNAs, snoRNAs, snRNAs, exRNAs, scaRNAs and the long ncRNAs such as Xist and HOTAIR.

Alternative splicing, or alternative RNA splicing, or differential splicing, is a regulated process during gene expression that results in a single gene coding for multiple proteins. In this process, particular exons of a gene may be included within or excluded from the final, processed messenger RNA (mRNA) produced from that gene. Consequently, the proteins translated from alternatively spliced mRNAs will contain differences in their amino acid sequence and, often, in their biological functions. Notably, alternative splicing allows the human genome to direct the synthesis of many more proteins than would be expected from its 20,000 protein-coding genes.
An Intergenic region (IGR) is a stretch of DNA sequences located between genes. Intergenic regions are a subset of noncoding DNA. Occasionally some intergenic DNA acts to control genes nearby, but most of it has no currently known function. It is one of the DNA sequences sometimes referred to as junk DNA, though it is only one phenomenon labeled such and in scientific studies today, the term is less used. Recently transcribed RNA from the DNA fragments in intergenic regions were known as "dark matter" or "dark matter transcripts".

In biology, a gene is a sequence of nucleotides in DNA or RNA that encodes the synthesis of a gene product, either RNA or protein.
Eukaryotic chromosome fine structure refers to the structure of sequences for eukaryotic chromosomes. Some fine sequences are included in more than one class, so the classification listed is not intended to be completely separate.
This glossary of genetics is a list of definitions of terms and concepts commonly used in the study of genetics and related disciplines in biology, including molecular biology and evolutionary biology. It is intended as introductory material for novices; for more specific and technical detail, see the article corresponding to each term. For related terms, see Glossary of evolutionary biology.
Long non-coding RNAs are a type of RNA, defined as being transcripts with lengths exceeding 200 nucleotides that are not translated into protein. This somewhat arbitrary limit distinguishes long ncRNAs from small non-coding RNAs such as microRNAs (miRNAs), small interfering RNAs (siRNAs), Piwi-interacting RNAs (piRNAs), small nucleolar RNAs (snoRNAs), and other short RNAs. Long intervening/intergenic noncoding RNAs (lincRNAs) are sequences of lncRNA which do not overlap protein-coding genes.
GENCODE is a scientific project in genome research and part of the ENCODE scale-up project.
A nested gene is a gene whose entire coding sequence lies within the bounds of a larger external gene. The coding sequence for a nested gene differs greatly from the coding sequence for its external host gene. Typically, nested genes and their host genes encode functionally unrelated proteins, and have different expression patterns in an organism.
Cryptic unstable transcripts (CUTs) are a subset of non-coding RNAs (ncRNAs) that are produced from intergenic and intragenic regions. CUTs were first observed in S. cerevisiae yeast models and are found in most eukaryotes. Some basic characteristics of CUTs include a length of around 200–800 base pairs, a 5' cap, poly-adenylated tail, and rapid degradation due to the combined activity of poly-adenylating polymerases and exosome complexes. CUT transcription occurs through RNA Polymerase II and initiates from nucleosome-depleted regions, often in an antisense orientation. To date, CUTs have a relatively uncharacterized function but have been implicated in a number of putative gene regulation and silencing pathways. Thousands of loci leading to the generation of CUTs have been described in the yeast genome. Additionally, stable uncharacterized transcripts, or SUTs, have also been detected in cells and bear many similarities to CUTs but are not degraded through the same pathways.
Epstein–Barr virus stable intronic-sequence RNAs (ebv-sisRNAs) are a class of non-coding RNAs generated by repeat introns in the Epstein–Barr virus. After EBERs 1 and 2, ebv-sisRNA-1 is the third most abundant EBV RNA generated during a highly oncogenic form of virus latency. Conservation of ebv-sisRNA sequence and secondary structure between EBV and other herpesviruses suggest shared functions in latent infection.
WormBase is an online biological database about the biology and genome of the nematode model organism Caenorhabditis elegans and contains information about other related nematodes. WormBase is used by the C. elegans research community both as an information resource and as a place to publish and distribute their results. The database is regularly updated with new versions being released every two months. WormBase is one of the organizations participating in the Generic Model Organism Database (GMOD) project.
A variant of uncertainsignificance (VUS) is an allele, or variant form of a gene, that has been identified through genetic testing but whose significance to the function or health of an organism is not known. Two related terms are "gene of uncertain significance" (GUS), which refers to a gene that has been identified through genome sequencing but whose connection to a human disease has not been established, and "insignificant mutation", referring to a gene variant that has no impact on the health or function of an organism. The term "variant' is favored in clinical practice over "mutation" because it can be used to describe an allele more precisely. When the variant has no impact on health, it is called a "benign variant". When it is associated with a disease, it is called a "pathogenic variant". A "pharmacogenomic variant" has an effect only when an individual takes a particular drug and therefore is neither benign nor pathogenic.

Short interspersed nuclear elements (SINEs) are non-autonomous, non-coding transposable elements (TEs) that are about 100 to 700 base pairs in length. They are a class of retrotransposons, DNA elements that amplify themselves throughout eukaryotic genomes, often through RNA intermediates. SINEs compose about 13% of the mammalian genome.

ANNOVAR is a bioinformatics software tool for the interpretation and prioritization of single nucleotide variants (SNVs), insertions, deletions, and copy number variants (CNVs) of a given genome. It has the ability to annotate human genomes hg18, hg19, hg38, and model organisms genomes such as: mouse, zebrafish, fruit fly, roundworm, yeast and many others. The annotations could be used to determine the functional consequences of the mutations on the genes and organisms, infer cytogenetic bands, report functional importance scores, and/or find variants in conserved regions. ANNOVAR along with SNP effect (SnpEFF) and Variant Effect Predictor (VEP) are three of the most commonly used variant annotation tools.
References
- 1 2 3 Gingeras, Thomas R. (2007). "Origin of phenotypes: Genes and transcripts". Genome Research. 17 (6): 682–690. doi: 10.1101/gr.6525007 . PMID 17567989.