PSP was first officially described by Richardson, Steele, and Olszewski in 1963 as a form of progressive parkinsonism.[4] However, the earliest known case presenting clinical features consistent with PSP, along with pathological confirmation, was reported in France in 1951.[5] Originally thought to be a more general type of atypical parkinsonism, PSP is now linked to distinct clinical phenotypes including PSP-Richardson's syndrome (PSP-RS), which is the most common sub-type of the disease.[6] As PSP advances to a fully symptomatic stage, many PSP subtypes eventually exhibit the clinical characteristics of PSP-RS.[3]
PSP, encompassing all its phenotypes, has a prevalence of 18 per 100,000, whereas PSP-RS affects approximately 5 to 7 per 100,000 individuals.[1][3] The first symptoms typically occur at 60–70 years of age. Males are slightly more likely to be affected than females.[1] No association has been found between PSP and any particular race, location, or occupation.[1]
Signs and symptoms
The initial symptoms in two-thirds of cases are loss of balance, lunging forward when mobilizing, fast walking, bumping into objects or people, and falls.[7][3][4]Dementia symptoms are also initially seen in about one in five cases.[8]
Later symptoms and signs can include, but do not necessarily include dementia (typically including loss of inhibition and ability to organize information), slurring of speech, difficulty swallowing, and difficulty moving the eyes, particularly in the vertical direction. The latter accounts for some of the falls experienced by these patients, as they find it difficult to look up or down.[9]
The visual symptoms are of particular importance in the diagnosis of this disorder. Patients typically complain of difficulty reading due to the inability to look downwards. The ophthalmoparesis experienced by these patients mainly concerns voluntary eye movement and the inability to make vertical saccades, which is often worse with downward saccades. Patients tend to have difficulty looking down (a downgaze palsy) followed by the addition of an upgaze palsy. This vertical gaze paresis will correct when the examiner passively rolls the patient's head up and down as part of a test for the oculocephalic reflex. Involuntary eye movement, as elicited by Bell's phenomenon, for instance, may be closer to normal.[10]
On close inspection, eye movements called "square-wave jerks" may be visible when the patient fixes gaze at distance. These are fine movements, that can be mistaken for nystagmus, except that they are saccadic in nature, with no smooth phase. Although healthy individuals also make square-wave jerk movements, PSP patients make slower square-wave jerk movements, with smaller vertical components.[11] Assessment of these square-wave jerks and diminished vertical saccades is especially useful for diagnosing progressive supranuclear palsy, because these movements set PSP patients apart from other parkinsonian patients.[11] Difficulties with convergence (convergence insufficiency), where the eyes come closer together while focusing on something near, like the pages of a book, is typical. Because the eyes have trouble coming together to focus at short distances, the patient may complain of diplopia (double vision) when reading.[9]
A characteristic facial appearance known as procerus sign, with a wide-eye stare, furrowing of forehead with a frowning expression, and deepening of other facial creases, is also diagnostic of PSP.[12]
Signs and symptoms of PSP-RS subtype
PSP-RS is characterized by a combination of motor, ocular, cognitive, and speech-related impairments, that typically emerge in early stages of the disease. Symptoms of PSP-RS usually begin after 60 and steadily progress over time.[3] Clinical symptoms of PSP-RS often include unexplained falls, unsteady gait, bradykinesia, apathy, disinhibition, cognitive dysfunction, difficulty planning or multitasking, slow speech, and impaired ocular movement.[3][4][13] PSP-RS is also characterized by unresponsiveness to dopamine therapies often prescribed for those with Parkinson's disease.[14] Patients can present initially with symptoms more characteristic of the PSP-Parkinson (PSP-P) subtype which is characterized by asymmetric rigidity, resting tremor, and are more responsive to dopamine therapies such as levadopa compared to PSP-RS.[15] Clinical and pathological differences between PSP-P and PRSP-RS occur within the first year of the disease, with individuals with PSP-RS exhibiting faster progression of symptoms and lower survival rates after diagnosis. Diagnostic criteria distinguish between probable and possible PSP-RS, as definitive diagnosis requires post-mortem neuropathological confirmation.[14] The NINDS-SPSP criteria define probable PSP-RS as requiring both vertical supranuclear gaze palsy and early postural instability with falls, while possible PSP-RS requires either vertical gaze palsy or slowed vertical saccades combined with early falls.[14][13] PSP-RS effects more males than females, with a male to female ratio of 1.8:1.[4] Symptom progression tends to occur more quickly in PSP-RS than other sub-types with the average disease duration being 5.9 years and an average age of death of 72.1 years.[4][10]
Motor symptoms in PSP-RS
One of the key features of PSP-RS that occurs within the first year of symptom onset, is early postural instability which often leads to unexplained falls.[6][14][16] Additionally, patients present with axial rigidity, which is characterized by stiffness in the neck and body.[14] This is often accompanied with bradykinesia which is slowness of movement. Although PSP-RS is often misdiagnosed as Parkinson's disease, tremors are uncommon in PSP-RS.[4] Motor symptoms are often symmetrical in PSP-RS with both sides of the body being affected.[17]
Ocular symptoms in PSP-RS
A defining feature of PSP-RS is vertical supranuclear gaze palsy, which is difficulty with voluntary downward gaze.[18] Vertical supranuclear gaze palsy, a symptom characterized by decreased velocity and amplitude of vertical eye movements (saccades) is often the prominent diagnostic feature of PSP-RS. Approximately 40% of patients with PSP-RS experiencing supranuclear gaze palsy, but it may not present until 3–4 years after disease onset.[3][18] Individuals with PSP-RS also display other ocular motor symptoms such as dry, red and sore eyes, blurred vision, and difficulty focusing.[10] They may also experience spontaneous and involuntary eye-lid closure or apraxia of the eyelid opening.[10]
Cognitive and behavioral symptoms in PSP-RS
Cognitive changes are frequent in PSP-RS compared to other PSP subtypes and include slowed thinking (bradyphrenia), executive dysfunction, and difficulty with planning or problem-solving.[14][16] Some patients exhibit apathy, emotional blunting, or involuntary episodes of laughing or crying unrelated to mood.[10] Some studies report that around half the individuals with PSP-RS develop these personality changes within 2-years of diagnosis.[10] Dementia is not typically a dominant feature early on but may develop in later stages.
Cause
The cause of PSP is unknown. Fewer than 1% of those with PSP have a family member with the same disorder. A variant in the MAPT gene for tau protein called the H1haplotype, located on chromosome 17 (rs1800547), has been linked to PSP.[19] Nearly all people with PSP received a copy of that variant from each parent, but this is true of about two-thirds of the general population. The H1 haplotype of the MAPT gene has been identified in approximately 94% of individuals with PSP, compared to around 78% in healthy adults.[20] Therefore, the H1 haplotype appears to be necessary but not sufficient to cause PSP. Other genes, as well as environmental toxins, are being investigated as other possible contributors to the cause of PSP.[21]
Additionally, the H2haplotype, combined with vascular dysfunction, seems to be a factor of vascular progressive supranuclear palsy.[22]
Genetic mechanisms in PSP-RS
Although there is a strong correlation between the H1 haplotype and PSP, the exact molecular mechanism remains unclear. Some individuals with PSP-RS have reported a family history of the disease, suggesting the possibility that genetic factors, such as the H1 haplotype, may contribute to inherited susceptibility in certain cases.[20][23] The PSP-RS subtype is known for a buildup of the 4R tau protein, which does not dissolve properly and forms insoluble aggregates in the brain. In healthy brains, there is typically a balanced ratio of 3-repeat (3R) and 4-repeat (4R) tau isoforms, resulting from the regulated inclusion or exclusion of exon 10 on the MAPT gene.[20] Inclusion of exon 10 promotes the production of 4R tau, while its exclusion favors 3R tau.[20][23] In PSP-RS, this balance is disrupted. Certain genetic variations in MAPT, most notably the H1/H1 haplotype, are associated with increased inclusion of exon 10, leading to elevated levels of 4R tau relative to 3R tau.[20] The resulting overproduction of 4R tau is believed to contribute to the pathological accumulation of tau aggregates observed in PSP-RS.
Risk factors
Risk factors for PSP are still being explored, but research has begun to uncover potential genetic, environmental, and biological contributors that may increase the likelihood of developing this condition. Associations between hypertension and increased risk for PSP development have been uncovered.[10] Additionally, ties to cerebrovascular disease and diabetes mellitus has been discovered, with type 2 diabetes being associated with increased brain atrophy and neurodegeneration.[10] Cerebrovascular disease may increase risk of vascular pathology and enhance disease burden through decreased blood flow and secondary injury.[10]
The image illustrates abnormal aggregation of 4-repeat (4R) tau protein (blue) disrupting microtubule structures (red), which impairs neuronal stability and function. Tau accumulation is a key pathological hallmark of PSP-RS and contributes to neurodegeneration.
The affected brain cells are both neurons and glial cells. The neurons display neurofibrillary tangles (NFTs), which are clumps of tau protein, a normal part of a brain cell's internal structural skeleton.[10] These tangles are often different from those seen in Alzheimer's disease, but may be structurally similar when they occur in the cerebral cortex.[25] Their chemical composition is usually different, however, and is similar to that of tangles seen in corticobasal degeneration.[26] Individuals with PSP typically exhibit reduced levels of total tau and phosphorylated tau in CSF compared to the elevated levels observed in Alzheimer's disease, but these levels are still higher than those found in healthy controls.[10] One biomarkers for PSP is neurofilament light chain (NfL), a non-specific marker of axonal damage.[10] NfL levels in the CSF of individuals with PSP are reported to be two to five times higher than those in healthy individuals or patients with Parkinson's disease, Parkinson's dementia, or dementia with Lewy bodies.[10] However, NfL levels are similar in PSP and other disorders like corticobasal syndrome (CBS) and MSA, limiting its utility for differential diagnosis among these conditions.[10] Still, NfL might help track how PSP gets worse over time.[10] Blood-based measurements of NfL have also shown promise, with plasma levels strongly correlating with CSF concentrations.[10]
Immunohistochemical image of a tau-positive tufted astrocyte in a brain sample from a patient with PSP-RS. Tufted astrocytes are a pathological hallmark of PSP and are characterized by abnormal accumulation of 4-repeat (4R) tau protein within astrocytic processes.
Tufts of tau protein in astrocytes, or tufted astrocytes, are also considered diagnostic. Tufted astrocytes are astrocytes that accumulate abnormally phosphorylated 4R tau in their proximal processes.[14] These astrocytic inclusions are considered a key distinguishing pathological marker of PSP-RS and are particularly concentrated in affected subcortical regions.[14][20] PSP is specifically associated with an increase in the deposition of 4R tau proteins, which differ from other tauopathies such as Alzheimer's disease, where both 3-repeat and 4-repeat tau are present.[14] Unlike globose NFTs, tau depositions may be more widespread in the cortex.[20][27]Lewy bodies are seen in some cases, but whether this is a variant or an independent co-existing process is not clear, and in some cases, PSP can coexist with corticobasal degeneration, Parkinson's, and/or Alzheimer's disease, particularly with older patients.[28][29][30][31][32] Additional pathological features include oligodendroglial coiled bodies, neuronal loss, and gliosis.[14][10] These changes contribute to the characteristic symptoms of the disease, such as postural instability (from midbrain atrophy), gaze palsy (due to involvement of vertical gaze centers), and cognitive decline (from frontal lobe degeneration).[14]
The principal areas of the brain affected are the:[10][14]
spinal cord, particularly the area where some control of the bladder and bowel resides
The progression of tau pathology in PSP is thought to follow a pattern that begins in the striatum and advances to the frontal and parietal lobes, then to the temporal and occipital lobes, and ultimately the brainstem.[20]
Tau pathology in PSP-RS is generally more severe than in other subtypes and frequently involves regions such as the basal ganglia, subthalamic nucleus, tectum, locus coeruleus, and dentate nucleus.[16] These differences are captured by the PSP-tau score, a semiquantitative 12-point scale used to assess the extent and distribution of tau pathology, with scores ranging from 0 (minimal involvement) to 12 (widespread and severe involvement).[16] While individuals with PSP-P typically have PSP-tau scores greater than 5, PSP-RS cases often present with higher scores, reflecting a greater overall tau burden in this subtype.[16]
Diagram of central cholinergic pathways that may be disrupted in PSP-RS. Degeneration in regions such as the pedunculopontine nucleus and nucleus basalis of Meynert can impair acetylcholine transmission to cortical and subcortical structures involved in memory, attention, and motor control.
Individuals with PSP-RS also exhibit early neuronal loss in the pedunculopontine tegmentum (PPT), an area of the brain responsible for producing acetylcholine, a neurotransmitter involved in memory, learning, and motor function.[39] The PPT sends cholinergic projections to several regions commonly affected by tau pathology in PSP, including the globus pallidus, substantia nigra, and pons.[39] Degeneration of cholinergic neurons in the PPT can also disrupt the function of other neurotransmitter systems, particularly dopamine, which plays a key role in motor regulation. In PSP-RS, dopaminergic neuronal loss has been partly attributed to reduced cholinergic input to the substantia nigra, further contributing to motor impairments characteristic of the disease.[39]
Diagnosis
Magnetic resonance imaging (MRI) is often used to diagnose PSP. MRI may show atrophy in the midbrain with preservation of the pons giving a "hummingbird" sign.[3][40]
Differential diagnosis
PSP is frequently misdiagnosed as Parkinson's disease because they both involve slowed movements and gait difficulty, with PSP being one of a collection of diseases referred to as Parkinson plus syndromes. Both Parkinson's and PSP have an onset in late middle age and involve slowing and rigidity of movement. However, several distinguishing features exist. Tremor is very common with Parkinson's, but rare with PSP. Speech and swallowing difficulties are more common and severe with PSP and the abnormal eye movements of PSP are essentially absent with PD.[41] A poor response to levodopa, along with symmetrical onset can also help differentiate PSP from PD.[42]
PSP can also be misdiagnosed as Alzheimer's disease because of the behavioral changes.[43]
PSP-RS is the most common subtype of PSP. In PSP-P features of Parkinson's Disease overlap with the clinical presentation of PSP and follows a more benign course. In both PSP-P and PSP- PAGF distribution of abnormal tau is relatively restricted to the brain stem. Frontal PSP initially presents with behavioral and cognitive symptoms, with or without ophthalmoparesis and then evolve into typical PSP.[12] The phenotypes of PSP-P and PSP-PAGF are sometimes referred as the "brain stem" variants of PSP, as opposed to the "cortical" variants which present with predominant cortical features, including PSP-CBS, PSP-bvFTD, and PSP-PNFA.[47]Cerebellar ataxia as the predominant early presenting feature is increasingly recognized as a very rare subtype of PSP (PSP-C) which is associated with severe neuronal loss with gliosis and higher densities of coiled bodies in the cerebellar dentate nucleus.[48]
The "humming-bird" imaging feature, indicative of midbrain degeneration, supports early diagnosis of both PSP-RS and PSP-P.[4][49] One commonly used quantitative measure is the midbrain-to-pons area ratio, which is calculated by measuring the cross-sectional areas of the midbrain and pons on midsagittal MRI images.[14]In healthy individuals, the midbrain and pons are proportionally sized, but in PSP-RS, the midbrain undergoes significant atrophy while the pons remains relatively spared.[14][13][49][50] As a result, the ratio is markedly reduced in PSP-RS, distinguishing it from other neurodegenerative conditions such as Parkinson's disease, multiple system atrophy (MSA), and even PSP-P, which typically exhibit less pronounced midbrain atrophy.[14][13][49][50] To improve diagnostic specificity, the magnetic resonance parkinsonism index (MRPI) incorporates the midbrain-to-pons area ratio along with measurements of the widths of the middle and superior cerebellar peduncles.[14] The MRPI has been shown to enhance discrimination between PSP-RS and other forms of atypical parkinsonism.[14] Additional MRI findings associated with PSP-RS may include third ventricle enlargement, thinning of the superior cerebellar peduncles, and frontal lobe atrophy, all of which are consistent with the underlying tau pathology seen in the disease.[14] Brain atrophy in PSP-RS is generally symmetrical, which may correspond to the bilateral presentation of motor and cognitive symptoms.[17]
PSP-RS is thought to follow a grading system that reflects the progressive nature of the disease.[20] In the early stages, atrophy affects midbrain structures such as the pons, and then progresses to involve the basal ganglia and pontine nuclei.[50] In later stages, the dentate gyrus of the hippocampus and the cerebral cortices exhibit cell death and atrophy.[50][49] PSP-RS is also associated with more widespread and severe cortical volume loss, particularly in the frontal pole, inferior frontal gyrus, globus pallidus, amygdala, and thalamus.[49] Additionally, diffusion tensor imaging (DTI) studies show significant reductions in fractional anisotropy, a measure of white matter integrity, in the body of the corpus callosum, superior cerebellar peduncles, dentatorubrothalamic tract, and anterior thalamic radiation, indicating extensive white matter disruption in PSP-RS.[51][50]
Positron emission tomography (PET) imaging has been used to detect tau accumulation in PSP-RS. The use of various tracers such as 18F-5105, 18F-FDDNP, 18F-THK523, 11C-PBB3 have shown increased binding in PSP-RS patients compared to healthy controls, reflecting elevated levels of 4R tau.[6] Brain scans called SPECT have been used to help diagnose different types of Parkinson-like conditions, including PSP-RS.[10] These scans show areas of reduced blood flow in the brain, but they aren't specific enough to clearly tell PSP-RS apart from other similar conditions like PSP-P or corticobasal syndrome (CBS).[10] Additionally, SPECT revealed greater perfusion deficits in PSP-RS than in PSP-P, although CBS showed the most frontal lobe deterioration overall.[10]
Differential diagnosis between PSP-subtypes
The table below summarizes key clinical, pathological, and imaging differences between the main subtypes of PSP, including PSP-RS, PSP-P, and other variant forms. While all subtypes share the underlying 4-repeat tau pathology characteristic of PSP, they differ in symptom onset, disease progression, anatomical vulnerability, and response to treatment. Patients with the Richardson variant of PSP tend to have an upright posture or arched back, as opposed to the stooped-forward posture of other Parkinsonian disorders, although PSP-Parkinsonism (see below) can demonstrate a stooped posture.[52] Early falls are also more common with PSP, especially with Richardson syndrome.[53]
Feature
PSP-RS
PSP-P
Other PSP Subtypes (e.g., PSP-SL, PSP-CBS, PSP-F, PSP-PG)
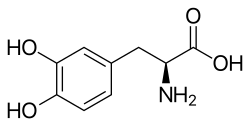
Chemical structure of levodopa (L-DOPA), a precursor to dopamine used in the treatment of Parkinsonian disorders, including PSP-RS. Levodopa crosses the blood–brain barrier and is converted to dopamine in the brain, temporarily alleviating motor symptoms.
Management is only supportive as a cure for PSP is unknown. PSP cases are often split into two subgroups, PSP-RS and PSP-P, where a short-term response to levodopa can be obtained.[10][56]Dyskinesia is an occasional but rare complication of treatment.[57] Other variants have been described.[58][59][60][61]Botox can be used to treat neck dystonia and blepharospasm, but this can aggravate dysphagia.[62]
Two studies have suggested that rivastigmine may help with cognitive aspects, but the authors of both studies have suggested that larger studies are needed.[63][64] There is some evidence from small-scale studies that the hypnotic zolpidem may improve motor function and eye movements.[65][66]
Current clinical trials focus on disease modifying treatments but there are currently no FDA approved disease modifying treatments for PSP-RS.[39] Recent trials have focused on therapies targeting the 4R tau accumulation in the brain of those with PSP-RS. These therapies include microtubule-stabilizing agents, tau gain-of-function therapies, and monoclonal antibodies.[10][39] These therapies are currently being tested in clinical trials.[10][39]
Rehabilitation
Patients with PSP usually seek or are referred to occupational therapy, speech-language pathology for motor speech changes (typically a spastic-ataxic dysarthria), and physical therapy for balance and gait problems with reports of frequent falls.[67] There has been research in the use of robot-assisted gait training.[68] Evidence-based approaches to rehabilitation in PSP are lacking and, currently the majority of research on the subject consists of case reports involving only a small number of patients.[69]
Case reports of rehabilitation programs for patients with PSP generally include limb-coordination activities, tilt-board balancing, gait training, strength training with progressive resistive exercises, and isokinetic exercises and stretching of the neck muscles.[67] While some case reports suggest that physiotherapy can offer improvements in balance and gait of patients with PSP, the results cannot be generalized across all PSP patients, as each case report followed only one or two patients.[67] The observations made from these case studies can be useful however, in helping to guide future research concerning the effectiveness of balance and gait training programs in the management of PSP.[39]
Individuals with PSP are often referred to occupational therapists to help manage their condition and to help enhance their independence. This may include being taught to use mobility aids.[70][71] Due to their tendency to fall backwards, the use of a walker, particularly one that can be weighted in the front, is recommended instead of a cane.[70] The use of an appropriate mobility aid helps to decrease the individual's risk of falls and makes them safer to ambulate independently in the community.[71] Due to their balance problems and irregular movements, individuals need to spend time learning how to safely transfer in their homes and in the community.[70] This may include rising from and sitting in chairs safely.[71]
Due to the progressive nature of this disease, all individuals eventually lose their ability to walk and will need to progress to using a wheelchair.[70] Severe dysphagia often follows, and at this point death is often a matter of months.[72]
Prognosis
No effective treatment or cure has been found for PSP, although some of the symptoms can respond to nonspecific measures. The poor prognosis is predominantly attributed to the serious impact this condition has on the quality of life.[7] The average age at symptoms onset is 63 and survival from onset averages seven years with a wide variance.[73]Pneumonia is a frequent cause of death, often caused by accidental aspiration of food particles.[74]
History
In 1877, Charcot described a 40-year-old woman who had rigid-akinetic parkinsonism, neck dystonia, dysarthria, and eye-movement problems. In 1951, Chavany and others reported the clinical and pathologic features of a 50-year-old man with a rigid and akinetic form of parkinsonism with postural instability, neck dystonia, dysarthria, and staring gaze.[5] In 1974, Albert and colleagues first described the unique frontal lobe cognitive changes of progressive supranuclear palsy—apathy, loss of spontaneity, slowing of thought processes, and loss of executive functions.[5][75]
Between 1877 and 1963, 22 well-documented case reports of PSP, although not described as a distinct disorder, had been identified in the literature of neurology.[76] Progressive supranuclear palsy was first described as a distinct disorder by neurologists John Steele, John Richardson, and Jerzy Olszewski in 1963.[77][78][79][80] They recognized the same clinical syndrome in eight patients, and described the autopsy findings in six of them.[78]
Society and culture
There are several organizations around the world that support PSP patients and the research into PSP and related diseases, such as corticobasal degeneration (CBD) and multiple system atrophy (MSA).
Canada: PSP Society of Canada, a federally registered non-profit organization which serves patients and families dealing with PSP, CBD and MSA, set up in 2017 through the help of CurePSP in the USA[81]
France: Association PSP France, a nonprofit patient association set up in 1996 through the help of PSPA in the UK. It also gives support to French speaking patients in Quebec, Morocco, Algeria, Belgium and Lebanon[82]
UK: PSPA, a national charity for information, patient support and research of PSP and CBD, set up in 1994[83][84]
Ireland: PSPAI, an organization which aims to increase public awareness of PSP[85]
US: CurePSP, a nonprofit organization for promoting awareness, care and research of PSP, CBD, MSA "and other prime of life neurodegenerative diseases"[86]
↑Amano N, Iwabuchi K, Yokoi S, Yagishita S, Itoh Y, Saitoh A, etal. (January 1989). "[The reappraisal study of the ultrastructure of Alzheimer's neurofibrillary tangles in three cases of progressive supranuclear palsy]". No to Shinkei = Brain and Nerve (in Japanese). 41 (1): 35–44. PMID2655673.
↑Kertesz A, Munoz D (2004). "Relationship between frontotemporal dementia and corticobasal degeneration/progressive supranuclear palsy". Dementia and Geriatric Cognitive Disorders. 17 (4): 282–6. doi:10.1159/000077155. PMID15178937. S2CID21017979.
↑Katsuse O, Iseki E, Arai T, Akiyama H, Togo T, Uchikado H, etal. (September 2003). "4-repeat tauopathy sharing pathological and biochemical features of corticobasal degeneration and progressive supranuclear palsy". Acta Neuropathologica. 106 (3): 251–60. doi:10.1007/s00401-003-0728-8. PMID12802605. S2CID20275104.
↑Hattori M, Hashizume Y, Yoshida M, Iwasaki Y, Hishikawa N, Ueda R, etal. (August 2003). "Distribution of astrocytic plaques in the corticobasal degeneration brain and comparison with tuft-shaped astrocytes in the progressive supranuclear palsy brain". Acta Neuropathologica. 106 (2): 143–9. doi:10.1007/s00401-003-0711-4. PMID12732936. S2CID25741692.
↑Komori T, Arai N, Oda M, Nakayama H, Mori H, Yagishita S, etal. (October 1998). "Astrocytic plaques and tufts of abnormal fibers do not coexist in corticobasal degeneration and progressive supranuclear palsy". Acta Neuropathologica. 96 (4): 401–8. doi:10.1007/s004010050911. PMID9797005. S2CID7265831.
↑Zhu MW, Wang LN, Li XH, Gui QP (April 2004). "[Glial abnormalities in progressive supranuclear palsy and corticobasal degeneration]" [Glial abnormalities in progressive supranuclear palsy and corticobasal degeneration]. Zhonghua Bing Li Xue Za Zhi = Chinese Journal of Pathology (in Chinese). 33 (2): 125–9. doi:10.3760/j.issn:0529-5807.2004.02.008. PMID15132848.
↑Wang LN, Zhu MW, Feng YQ, Wang JH (June 2006). "Pick's disease with Pick bodies combined with progressive supranuclear palsy without tuft-shaped astrocytes: a clinical, neuroradiologic and pathological study of an autopsied case". Neuropathology. 26 (3): 222–30. doi:10.1111/j.1440-1789.2006.00671.x. PMID16771179. S2CID25562683.
↑Dickson DW, Ahmed Z, Algom AA, Tsuboi Y, Josephs KA (August 2010). "Neuropathology of variants of progressive supranuclear palsy". Current Opinion in Neurology. 23 (4): 394–400. doi:10.1097/WCO.0b013e32833be924. PMID20610990.
↑Kanazawa M, Tada M, Onodera O, Takahashi H, Nishizawa M, Shimohata T (2013). "Early clinical features of patients with progressive supranuclear palsy with predominant cerebellar ataxia". Parkinsonism Relat Disord. 19 (12): 1149–51. doi:10.1016/j.parkreldis.2013.07.019. PMID23916652.
↑Nijboer H, Dautzenberg PL (June 2009). "[Progressive supranucleair palsy: acetylcholineeserase-inhibitor a possible therapy?]". Tijdschrift voor Gerontologie en Geriatrie. 40 (3): 133–7. doi:10.1007/BF03079574. PMID19731749. S2CID140525754.
↑Liepelt I, Gaenslen A, Godau J, Di Santo A, Schweitzer KJ, Gasser T, etal. (January 2010). "Rivastigmine for the treatment of dementia in patients with progressive supranuclear palsy: Clinical observations as a basis for power calculations and safety analysis". Alzheimer's & Dementia. 6 (1): 70–4. doi:10.1016/j.jalz.2009.04.1231. PMID20129321. S2CID33349776.
12Richardson JC, Steele J, Olszewski J (1963). "Supranuclear Ophthalmoplegia, Pseudobulbar Palsy, Nuchal Dystonia and Dementia. A Clinical Report on Eight Cases of 'heterogenous System Degeneration'". Transactions of the American Neurological Association. 88: 25–9. PMID14272249.
This page is based on this Wikipedia article Text is available under the CC BY-SA 4.0 license; additional terms may apply. Images, videos and audio are available under their respective licenses.