
Spinal muscular atrophies (SMAs) are a genetically and clinically heterogeneous group of rare debilitating disorders characterised by the degeneration of lower motor neurons and subsequent atrophy (wasting) of various muscle groups in the body. While some SMAs lead to early infant death, other diseases of this group permit normal adult life with only mild weakness.

Arthrogryposis (AMC) describes congenital joint contracture in two or more areas of the body. It derives its name from Greek, literally meaning 'curving of joints'.
Hypotonia is a state of low muscle tone, often involving reduced muscle strength. Hypotonia is not a specific medical disorder, but a potential manifestation of many different diseases and disorders that affect motor nerve control by the brain or muscle strength. Hypotonia is a lack of resistance to passive movement, whereas muscle weakness results in impaired active movement. Central hypotonia originates from the central nervous system, while peripheral hypotonia is related to problems within the spinal cord, peripheral nerves and/or skeletal muscles. Severe hypotonia in infancy is commonly known as floppy baby syndrome. Recognizing hypotonia, even in early infancy, is usually relatively straightforward, but diagnosing the underlying cause can be difficult and often unsuccessful. The long-term effects of hypotonia on a child's development and later life depend primarily on the severity of the muscle weakness and the nature of the cause. Some disorders have a specific treatment but the principal treatment for most hypotonia of idiopathic or neurologic cause is physical therapy and/or occupational therapy for remediation.
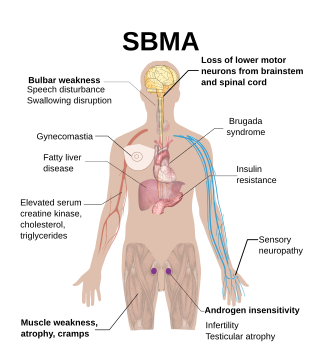
Spinal and bulbar muscular atrophy (SBMA), popularly known as Kennedy's disease, is a rare, adult-onset, X-linked recessive lower motor neuron disease caused by trinucleotide CAG repeat expansions in exon 1 of the androgen receptor (AR) gene, which results in both loss of AR function and toxic gain of function.
Hereditary inclusion body myopathies (HIBM) are a group of rare genetic disorders which have different symptoms. Generally, they are neuromuscular disorders characterized by muscle weakness developing in young adults. Hereditary inclusion body myopathies comprise both autosomal recessive and autosomal dominant muscle disorders that have a variable expression (phenotype) in individuals, but all share similar structural features in the muscles.

Centronuclear myopathies (CNM) are a group of congenital myopathies where cell nuclei are abnormally located in the center of muscle cells instead of their normal location at the periphery.

Congenital muscular dystrophies are autosomal recessively-inherited muscle diseases. They are a group of heterogeneous disorders characterized by muscle weakness which is present at birth and the different changes on muscle biopsy that ranges from myopathic to overtly dystrophic due to the age at which the biopsy takes place.

Emery–Dreifuss muscular dystrophy (EDMD) is a type of muscular dystrophy, a group of heritable diseases that cause progressive impairment of muscles. EDMD affects muscles used for movement, causing atrophy, weakness and contractures. It almost always affects the heart, causing abnormal rhythms, heart failure, or sudden cardiac death. It is rare, affecting 0.39 per 100,000 people. It is named after Alan Eglin H. Emery and Fritz E. Dreifuss.
Spinal muscular atrophy (SMA) is a rare neuromuscular disorder that results in the loss of motor neurons and progressive muscle wasting. It is usually diagnosed in infancy or early childhood and if left untreated it is the most common genetic cause of infant death. It may also appear later in life and then have a milder course of the disease. The common feature is progressive weakness of voluntary muscles, with arm, leg and respiratory muscles being affected first. Associated problems may include poor head control, difficulties swallowing, scoliosis, and joint contractures.

Bethlem myopathy is predominantly an autosomal dominant myopathy, classified as a congenital form of muscular dystrophy. There are two forms of Bethlem myopathy.

X-linked spinal muscular atrophy type 2, also known as arthrogryposis multiplex congenita X-linked type 1 (AMCX1), is a rare neurological disorder involving death of motor neurons in the anterior horn of spinal cord resulting in generalised muscle wasting (atrophy). The disease is caused by a mutation in UBA1 gene and is passed in an X-linked recessive manner by carrier mothers to affected sons.
Distal spinal muscular atrophy type 1 (DSMA1), also known as spinal muscular atrophy with respiratory distress type 1 (SMARD1), is a rare neuromuscular disorder involving death of motor neurons in the spinal cord which leads to a generalised progressive atrophy of body muscles.
Cerebral dysgenesis–neuropathy–ichthyosis–keratoderma syndrome is a neurocutaneous condition caused by mutation in the SNAP29 gene.
Spinal muscular atrophy with progressive myoclonic epilepsy (SMA-PME), sometimes called Jankovic–Rivera syndrome, is a very rare neurodegenerative disease whose symptoms include slowly progressive muscle (atrophy), predominantly affecting proximal muscles, combined with denervation and myoclonic seizures. Only 12 known human families are described in scientific literature to have SMA-PME.
Parastremmatic dwarfism is a rare bone disease that features severe dwarfism, thoracic kyphosis, a distortion and twisting of the limbs, contractures of the large joints, malformations of the vertebrae and pelvis, and incontinence. The disease was first reported in 1970 by Leonard Langer and associates; they used the term parastremmatic from the Greek parastremma, or distorted limbs, to describe it. On X-rays, the disease is distinguished by a "flocky" or lace-like appearance to the bones. The disease is congenital, which means it is apparent at birth. It is caused by a mutation in the TRPV4 gene, located on chromosome 12 in humans. The disease is inherited in an autosomal dominant manner.

Monomelic amyotrophy (MMA) is a rare motor neuron disease first described in 1959 in Japan. Its symptoms usually appear about two years after adolescent growth spurt and is significantly more common in males, with an average age of onset between 15 and 25 years. MMA is reported most frequently in Asia but has a global distribution. It is typically marked by insidious onset of muscle atrophy of an upper limb, which plateaus after two to five years from which it neither improves nor worsens. There is no pain or sensory loss associated with MMA. MMA is not believed to be hereditary.
Jokela type spinal muscular atrophy (SMAJ), also known as late-onset spinal motor neuronopathy (LOSMoN), is an ultra-rare neuromuscular disorder characterized by muscle twitches and cramps. The symptoms appear in adulthood and gradually progress. The disease is caused by a mutation in the CHCHD10 gene and is inherited in an autosomal dominant pattern. It was first described by the Finnish neurologist Manu Jokela in 2011.

Charlotte Jane Sumner is an American neurologist. She is a professor in the Departments of Neurology and Neuroscience at Johns Hopkins School of Medicine. Dr. Sumner cares for patients with genetically mediated neuromuscular diseases and directs a laboratory focused on developing treatments for these diseases. She co-directs the Johns Hopkins Muscular Dystrophy Association Care Center, the Spinal Muscular Atrophy (SMA), and the Charcot-Marie-Tooth (CMT) clinics, which deliver multidisciplinary clinical care, engage in international natural history studies, and provide cutting edge therapeutics.

Autosomal recessive axonal neuropathy with neuromyotonia, also known as Gamstorp-Wohlfart syndrome, is a rare hereditary disorder which is characterized by progressive poly-neuropathy, neuromyotonia, myokymia, pseudo-myotonia, hand-foot contractures, and abnormal neuro-myotonic/myokimic activity visible on needle EMG. According to OMIM, around 52 cases have been reported in medical literature However; new cases have been reported since 2014.
