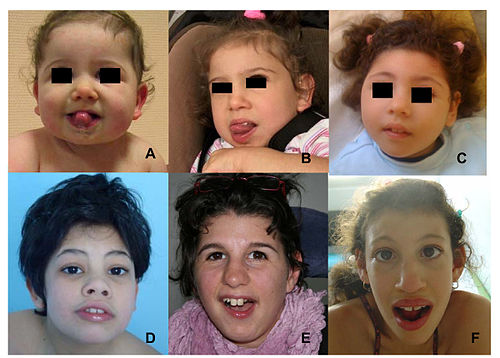
 Facial features (dysmorphism) of patients with one form of pontocerebellar hypoplasia due to mutations in the CASK gene. A and B: patient at 1 year (A) and 4 years (B). C: patient, 18 months. D: patient, 13 years. E: patient, 13 years. F: patient, 12 years. Note minor facial dysmorphism: round face, small chin, well-drawn eyebrows in the younger patients; longer face, high and large nasal bridge, long nose, protruding maxilla, in the older patients.
Facial features (dysmorphism) of patients with one form of pontocerebellar hypoplasia due to mutations in the CASK gene. A and B: patient at 1 year (A) and 4 years (B). C: patient, 18 months. D: patient, 13 years. E: patient, 13 years. F: patient, 12 years. Note minor facial dysmorphism: round face, small chin, well-drawn eyebrows in the younger patients; longer face, high and large nasal bridge, long nose, protruding maxilla, in the older patients. Magnetic resonance imaging (MRI) examples of patients with pontocerebellar hypoplasia with CASK mutations. A. Sagittal images showing different degrees of hypoplasia (incomplete formation) of the pons and vermis (parts of the brain). Numbers represent different patients. [3]
Magnetic resonance imaging (MRI) examples of patients with pontocerebellar hypoplasia with CASK mutations. A. Sagittal images showing different degrees of hypoplasia (incomplete formation) of the pons and vermis (parts of the brain). Numbers represent different patients. [3]
| Pontocerebellar hypoplasia | |
|---|---|
| Other names | Non-syndromic pontocerebellar hypoplasia |
 | |
| Pontocerebellar hypoplasia is inherited in an autosomal recessive manner. | |
| Specialty | Neurology |
| Symptoms | Intellectual disability, movement problems |
| Treatment | Unknown |
Pontocerebellar hypoplasia (PCH) is a heterogeneous group of rare neurodegenerative disorders caused by genetic mutations and characterised by progressive atrophy of various parts of the brain such as the cerebellum or brainstem (particularly the pons). [1] Where known, these disorders are inherited in an autosomal recessive fashion. There is no known cure for PCH. [2]

