Neuromyotonia (NMT) is a form of peripheral nerve hyperexcitability that causes spontaneous muscular activity resulting from repetitive motor unit action potentials of peripheral origin. NMT along with Morvan's syndrome are the most severe types in the Peripheral Nerve Hyperexciteability spectrum. Example of two more common and less severe syndromes in the spectrum are Cramp Fasciculation Syndrome and Benign Fasciculation Syndrome. NMT can have both hereditary and acquired forms. The prevalence of NMT is unknown.

Encephalitis is inflammation of the brain. The severity can be variable with symptoms including reduction or alteration in consciousness, headache, fever, confusion, a stiff neck, and vomiting. Complications may include seizures, hallucinations, trouble speaking, memory problems, and problems with hearing.
Morvan's syndrome is a rare, life-threatening autoimmune disease named after the nineteenth century French physician Augustin Marie Morvan. "La chorée fibrillaire" was first coined by Morvan in 1890 when describing patients with multiple, irregular contractions of the long muscles, cramping, weakness, pruritus, hyperhidrosis, insomnia and delirium. It normally presents with a slow insidious onset over months to years. Approximately 90% of cases spontaneously go into remission, while the other 10% of cases lead to death.
Opsoclonus myoclonus syndrome (OMS), also known as opsoclonus-myoclonus-ataxia (OMA), is a rare neurological disorder of unknown cause which appears to be the result of an autoimmune process involving the nervous system. It is an extremely rare condition, affecting as few as 1 in 10,000,000 people per year. It affects 2 to 3% of children with neuroblastoma and has been reported to occur with celiac disease and diseases of neurologic and autonomic dysfunction.
Neuromyelitis optica spectrum disorders (NMOSD), including neuromyelitis optica (NMO), are autoimmune diseases characterized by acute inflammation of the optic nerve and the spinal cord (myelitis). Episodes of ON and myelitis can be simultaneous or successive. A relapsing disease course is common, especially in untreated patients. In more than 80% of cases, NMO is caused by immunoglobulin G autoantibodies to aquaporin 4 (anti-AQP4), the most abundant water channel protein in the central nervous system. A subset of anti-AQP4-negative cases is associated with antibodies against myelin oligodendrocyte glycoprotein (anti-MOG). Rarely, NMO may occur in the context of other autoimmune diseases or infectious diseases. In some cases, the etiology remains unknown.

Stiff-person syndrome (SPS), also known as stiff-man syndrome, is a rare neurologic disorder of unclear cause characterized by progressive muscular rigidity and stiffness. The stiffness primarily affects the truncal muscles and is superimposed by spasms, resulting in postural deformities. Chronic pain, impaired mobility, and lumbar hyperlordosis are common symptoms.

Viral encephalitis is inflammation of the brain parenchyma, called encephalitis, by a virus. The different forms of viral encephalitis are called viral encephalitides. It is the most common type of encephalitis and often occurs with viral meningitis. Encephalitic viruses first cause infection and replicate outside of the central nervous system (CNS), most reaching the CNS through the circulatory system and a minority from nerve endings toward the CNS. Once in the brain, the virus and the host's inflammatory response disrupt neural function, leading to illness and complications, many of which frequently are neurological in nature, such as impaired motor skills and altered behavior.
Paraneoplastic cerebellar degeneration (PCD) is a paraneoplastic syndrome associated with a broad variety of tumors including lung cancer, ovarian cancer, breast cancer, Hodgkin’s lymphoma and others. PCD is a rare condition that occurs in less than 1% of cancer patients.
Inflammatory demyelinating diseases (IDDs), sometimes called Idiopathic (IIDDs) due to the unknown etiology of some of them, are a heterogenous group of demyelinating diseases - conditions that cause damage to myelin, the protective sheath of nerve fibers - that occur against the background of an acute or chronic inflammatory process. IDDs share characteristics with and are often grouped together under Multiple Sclerosis. They are sometimes considered different diseases from Multiple Sclerosis, but considered by others to form a spectrum differing only in terms of chronicity, severity, and clinical course.
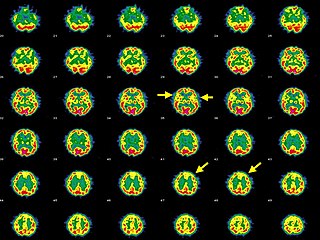
Hashimoto's encephalopathy, also known as steroid-responsive encephalopathy associated with autoimmune thyroiditis (SREAT), is a neurological condition characterized by encephalopathy, thyroid autoimmunity, and good clinical response to corticosteroids. It is associated with Hashimoto's thyroiditis, and was first described in 1966. It is sometimes referred to as a neuroendocrine disorder, although the condition's relationship to the endocrine system is widely disputed. It is recognized as a rare disease by the NIH Genetic and Rare Diseases Information Center.
A paraneoplastic syndrome is a syndrome that is the consequence of a tumor in the body. It is specifically due to the production of chemical signaling molecules by tumor cells or by an immune response against the tumor. Unlike a mass effect, it is not due to the local presence of cancer cells.
Anti-glutamate receptor antibodies are autoantibodies detected in serum and/or cerebrospinal fluid samples of a variety of disorders such as encephalitis, epilepsy and ataxia. Clinical and experimental studies starting around the year 2000 suggest that these antibodies are not simply epiphenomena and are involved in autoimmune disease pathogenesis.

Paraneoplastic antigen Ma2 is a protein that in humans is encoded by the PNMA2 gene.

Tumefactive multiple sclerosis is a condition in which the central nervous system of a person has multiple demyelinating lesions with atypical characteristics for those of standard multiple sclerosis (MS). It is called tumefactive as the lesions are "tumor-like" and they mimic tumors clinically, radiologically and sometimes pathologically.
Paraneoplastic pemphigus (PNP) is an autoimmune disorder stemming from an underlying tumor. It is hypothesized that antigens associated with the tumor trigger an immune response resulting in blistering of the skin and mucous membranes.

Anti-NMDA receptor encephalitis is a type of brain inflammation caused by antibodies. Early symptoms may include fever, headache, and feeling tired. This is then typically followed by psychosis which presents with false beliefs (delusions) and seeing or hearing things that others do not see or hear (hallucinations). People are also often agitated or confused. Over time, seizures, decreased breathing, and blood pressure and heart rate variability typically occur. In some cases, patients may develop catatonia.
Bickerstaff brainstem encephalitis is a rare inflammatory disorder of the central nervous system, first described by Edwin Bickerstaff in 1951. It may also affect the peripheral nervous system, and has features in common with both Miller Fisher syndrome and Guillain–Barré syndrome.

Autoimmune encephalitis (AIE) is a type of encephalitis, and one of the most common causes of noninfectious encephalitis. It can be triggered by tumors, infections, or it may be cryptogenic. The neurological manifestations can be either acute or subacute and usually develop within six weeks. The clinical manifestations include behavioral and psychiatric symptoms, autonomic disturbances, movement disorders, and seizures.
Anti-VGKC-complex encephalitis are caused by antibodies against the voltage gated potassium channel-complex (VGKC-complex) and are implicated in several autoimmune conditions including limbic encephalitis, epilepsy and neuromyotonia.
Anti-Hu associated encephalitis, also known as Anti-ANNA1 associated encephalitis, is an uncommon form of brain inflammation that is associated with an underlying cancer. It can cause psychiatric symptoms such as depression, anxiety, and hallucinations. It can also produce neurological symptoms such as confusion, memory loss, weakness, sensory loss, pain, seizures, and problems coordinating the movement of the body.
