Related Research Articles
A microsatellite is a tract of repetitive DNA in which certain DNA motifs are repeated, typically 5–50 times. Microsatellites occur at thousands of locations within an organism's genome. They have a higher mutation rate than other areas of DNA leading to high genetic diversity. Microsatellites are often referred to as short tandem repeats (STRs) by forensic geneticists and in genetic genealogy, or as simple sequence repeats (SSRs) by plant geneticists.
Repeated sequences are patterns of nucleic acids that occur in multiple copies throughout the genome. Repetitive DNA was first detected because of its rapid re-association kinetics. In many organisms, a significant fraction of the genomic DNA is highly repetitive, with over two-thirds of the sequence consisting of repetitive elements in humans.

A frameshift mutation is a genetic mutation caused by indels of a number of nucleotides in a DNA sequence that is not divisible by three. Due to the triplet nature of gene expression by codons, the insertion or deletion can change the reading frame, resulting in a completely different translation from the original. The earlier in the sequence the deletion or insertion occurs, the more altered the protein. A frameshift mutation is not the same as a single-nucleotide polymorphism in which a nucleotide is replaced, rather than inserted or deleted. A frameshift mutation will in general cause the reading of the codons after the mutation to code for different amino acids. The frameshift mutation will also alter the first stop codon encountered in the sequence. The polypeptide being created could be abnormally short or abnormally long, and will most likely not be functional.
In genetics, anticipation is a phenomenon whereby as a genetic disorder is passed on to the next generation, the symptoms of the genetic disorder become apparent at an earlier age with each generation. In most cases, an increase in the severity of symptoms is also noted. Anticipation is common in trinucleotide repeat disorders, such as Huntington's disease and myotonic dystrophy, where a dynamic mutation in DNA occurs. All of these diseases have neurological symptoms. Prior to the understanding of the genetic mechanism for anticipation, it was debated whether anticipation was a true biological phenomenon or whether the earlier age of diagnosis was related to heightened awareness of disease symptoms within a family.

Spinocerebellar ataxia (SCA) is a progressive, degenerative, genetic disease with multiple types, each of which could be considered a neurological condition in its own right. An estimated 150,000 people in the United States have a diagnosis of spinocerebellar ataxia at any given time. SCA is hereditary, progressive, degenerative, and often fatal. There is no known effective treatment or cure. SCA can affect anyone of any age. The disease is caused by either a recessive or dominant gene. In many cases people are not aware that they carry a relevant gene until they have children who begin to show signs of having the disorder.
Trinucleotide repeat disorders, also known as microsatellite expansion diseases, are a set of over 50 genetic disorders caused by trinucleotide repeat expansion, a kind of mutation in which repeats of three nucleotides increase in copy numbers until they cross a threshold above which they become unstable. Depending on its location, the unstable trinucleotide repeat may cause defects in a protein encoded by a gene; change the regulation of gene expression; produce a toxic RNA, or lead to chromosome instability. In general, the larger the expansion the faster the onset of disease, and the more severe the disease becomes.

Huntingtin(Htt) is the protein coded for by the HTT gene, also known as the IT15 gene. Mutated HTT is the cause of Huntington's disease (HD), and has been investigated for this role and also for its involvement in long-term memory storage.
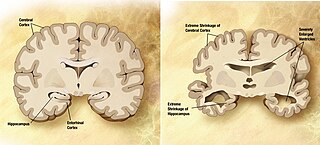
Neurodegeneration is the progressive loss of structure or function of neurons, which may ultimately involve cell death. Many neurodegenerative diseases—such as amyotrophic lateral sclerosis, multiple sclerosis, Parkinson's disease, Alzheimer's disease, Huntington's disease, and prion diseases—occur as a result of neurodegenerative processes. Neurodegeneration can be found in the brain at many different levels of neuronal circuitry, ranging from molecular to systemic. Because there is no known way to reverse the progressive degeneration of neurons, these diseases are considered to be incurable; however research has shown that the two major contributing factors to neurodegeneration are oxidative stress and inflammation. Biomedical research has revealed many similarities between these diseases at the sub-cellular level, including atypical protein assemblies and induced cell death. These similarities suggest that therapeutic advances against one neurodegenerative disease might ameliorate other diseases as well.

Slipped strand mispairing (SSM),, is a mutation process which occurs during DNA replication. It involves denaturation and displacement of the DNA strands, resulting in mispairing of the complementary bases. Slipped strand mispairing is one explanation for the origin and evolution of repetitive DNA sequences.
A trinucleotide repeat expansion, also known as a triplet repeat expansion, is the DNA mutation responsible for causing any type of disorder categorized as a trinucleotide repeat disorder. These are labelled in dynamical genetics as dynamic mutations. Triplet expansion is caused by slippage during DNA replication, also known as "copy choice" DNA replication. Due to the repetitive nature of the DNA sequence in these regions, 'loop out' structures may form during DNA replication while maintaining complementary base pairing between the parent strand and daughter strand being synthesized. If the loop out structure is formed from the sequence on the daughter strand this will result in an increase in the number of repeats. However, if the loop out structure is formed on the parent strand, a decrease in the number of repeats occurs. It appears that expansion of these repeats is more common than reduction. Generally, the larger the expansion the more likely they are to cause disease or increase the severity of disease. Other proposed mechanisms for expansion and reduction involve the interaction of RNA and DNA molecules.

Ataxin-1 is a DNA-binding protein which in humans is encoded by the ATXN1 gene.
Ataxin 7 (ATXN7) is a protein of the SCA7 gene, which contains 892 amino acids with an expandable poly(Q) region close to the N-terminus. The expandable poly(Q) motif region in the protein contributes crucially to spinocerebellar ataxia (SCA) pathogenesis by the induction of intranuclear inclusion bodies. ATXN7 is associated with both olivopontocerebellar atrophy type 3 (OPCA3) and spinocerebellar ataxia type 7 (SCA7).

Spinocerebellar ataxia type 6 (SCA6) is a rare, late-onset, autosomal dominant disorder, which, like other types of SCA, is characterized by dysarthria, oculomotor disorders, peripheral neuropathy, and ataxia of the gait, stance, and limbs due to cerebellar dysfunction. Unlike other types, SCA 6 is not fatal. This cerebellar function is permanent and progressive, differentiating it from episodic ataxia type 2 (EA2) where said dysfunction is episodic. In some SCA6 families, some members show these classic signs of SCA6 while others show signs more similar to EA2, suggesting that there is some phenotypic overlap between the two disorders. SCA6 is caused by mutations in CACNA1A, a gene encoding a calcium channel α subunit. These mutations tend to be trinucleotide repeats of CAG, leading to the production of mutant proteins containing stretches of 20 or more consecutive glutamine residues; these proteins have an increased tendency to form intracellular agglomerations. Unlike many other polyglutamine expansion disorders expansion length is not a determining factor for the age that symptoms present.
In genetics, a dynamic mutation is an unstable heritable element where the probability of expression of a mutant phenotype is a function of the number of copies of the mutation. That is, the replication product (progeny) of a dynamic mutation has a different likelihood of mutation than its predecessor. These mutations, typically short sequences repeated many times, give rise to numerous known diseases, including the trinucleotide repeat disorders.

Atrophin-1 is a protein that in humans is encoded by the ATN1 gene. The encoded protein includes a serine repeat and a region of alternating acidic and basic amino acids, as well as the variable glutamine repeat. The function of Atrophin-1 has not yet been determined. There is evidence provided by studies of Atrophin-1 in animals to suggest it acts as a transcriptional co-repressor. Atrophin-1 can be found in the nuclear and cytoplasmic compartments of neurons. It is expressed in nervous tissue.
Ataxin-2 is a protein that in humans is encoded by the ATXN2 gene. Mutations in ATXN2 cause spinocerebellar ataxia type 2 (SCA2).

Ataxin-3 is a protein that in humans is encoded by the ATXN3 gene.
Dentatorubral–pallidoluysian atrophy (DRPLA) is an autosomal dominant spinocerebellar degeneration caused by an expansion of a CAG repeat encoding a polyglutamine tract in the atrophin-1 protein. It is also known as Haw River Syndrome and Naito–Oyanagi disease. Although this condition was perhaps first described by Smith et al. in 1958, and several sporadic cases have been reported from Western countries, this disorder seems to be very rare except in Japan.
Autosomal dominant cerebellar ataxia (ADCA) is a form of spinocerebellar ataxia inherited in an autosomal dominant manner. ADCA is a genetically inherited condition that causes deterioration of the nervous system leading to disorder and a decrease or loss of function to regions of the body.
Spinocerebellar ataxia type 1 (SCA1) is a rare autosomal dominant disorder, which, like other spinocerebellar ataxias, is characterized by neurological symptoms including dysarthria, hypermetric saccades, and ataxia of gait and stance. This cerebellar dysfunction is progressive and permanent. First onset of symptoms is normally between 30 and 40 years of age, though juvenile onset can occur. Death typically occurs within 10 to 30 years from onset.
References
- ↑ Usdin K, House NC, Freudenreich CH (2015). "Repeat instability during DNA repair: Insights from model systems". Crit. Rev. Biochem. Mol. Biol. 50 (2): 142–67. doi:10.3109/10409238.2014.999192. PMC 4454471 . PMID 25608779.
- ↑ Laura Bonetta, Polyglutamine Diseases: A Devastating Genetic Stutter Archived 2013-05-07 at the Wayback Machine , Howard Hughes Medical Institute. Retrieved 30 December 2008.
- ↑ Wharton KA, Yedvobnick B, Finnerty VG, Artavanis-Tsakonas S (1985). "opa: a novel family of transcribed repeats shared by the Notch locus and other developmentally regulated loci in D. melanogaster ". Cell. 40 (1): 55–62. doi:10.1016/0092-8674(85)90308-3. PMID 2981631. S2CID 9951524.
- ↑ Rice C, Beekman D, Liu L, Erives A (2015). "The Nature, Extent, and Consequences of Genetic Variation in the opa Repeats of Notch in Drosophila". G3: Genes, Genomes, Genetics. 5 (15): 2405–2419. doi:10.1534/g3.115.021659. PMC 4632060 . PMID 26362765.