
Proteasomes are protein complexes which degrade ubiquitin-tagged proteins by proteolysis, a chemical reaction that breaks peptide bonds. Enzymes that help such reactions are called proteases.

In molecular biology, molecular chaperones are proteins that assist the conformational folding or unfolding of large proteins or macromolecular protein complexes. There are a number of classes of molecular chaperones, all of which function to assist large proteins in proper protein folding during or after synthesis, and after partial denaturation. Chaperones are also involved in the translocation of proteins for proteolysis.
AAAproteins are a large group of protein family sharing a common conserved module of approximately 230 amino acid residues. This is a large, functionally diverse protein family belonging to the AAA+ protein superfamily of ring-shaped P-loop NTPases, which exert their activity through the energy-dependent remodeling or translocation of macromolecules.
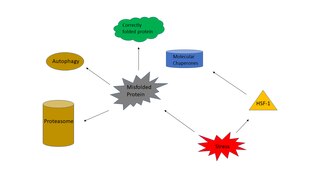
The heat shock response (HSR) is a cell stress response that increases the number of molecular chaperones to combat the negative effects on proteins caused by stressors such as increased temperatures, oxidative stress, and heavy metals. In a normal cell, proteostasis must be maintained because proteins are the main functional units of the cell. Many proteins take on a defined configuration in a process known as protein folding in order to perform their biological functions. If these structures are altered, critical processes could be affected, leading to cell damage or death. The heat shock response can be employed under stress to induce the expression of heat shock proteins (HSP), many of which are molecular chaperones, that help prevent or reverse protein misfolding and provide an environment for proper folding.
Inclusion bodies are aggregates of specific types of protein found in neurons, and a number of tissue cells including red blood cells, bacteria, viruses, and plants. Inclusion bodies of aggregations of multiple proteins are also found in muscle cells affected by inclusion body myositis and hereditary inclusion body myopathy.

A fungal prion is a prion that infects hosts which are fungi. Fungal prions are naturally occurring proteins that can switch between multiple, structurally distinct conformations, at least one of which is self-propagating and transmissible to other prions. This transmission of protein state represents an epigenetic phenomenon where information is encoded in the protein structure itself, instead of in nucleic acids. Several prion-forming proteins have been identified in fungi, primarily in the yeast Saccharomyces cerevisiae. These fungal prions are generally considered benign, and in some cases even confer a selectable advantage to the organism.

A viroplasm, sometimes called "virus factory" or "virus inclusion", is an inclusion body in a cell where viral replication and assembly occurs. They may be thought of as viral factories in the cell. There are many viroplasms in one infected cell, where they appear dense to electron microscopy. Very little is understood about the mechanism of viroplasm formation.

Endoplasmic-reticulum-associated protein degradation (ERAD) designates a cellular pathway which targets misfolded proteins of the endoplasmic reticulum for ubiquitination and subsequent degradation by a protein-degrading complex, called the proteasome.
In eukaryotic cells, an aggresome refers to an aggregation of misfolded proteins in the cell, formed when the protein degradation system of the cell is overwhelmed. Aggresome formation is a highly regulated process that possibly serves to organize misfolded proteins into a single location.
The unfolded protein response (UPR) is a cellular stress response related to the endoplasmic reticulum (ER) stress. It has been found to be conserved between mammalian species, as well as yeast and worm organisms.

26S protease regulatory subunit S10B, also known as 26S proteasome AAA-ATPase subunit Rpt4, is an enzyme that in humans is encoded by the PSMC6 gene. This protein is one of the 19 essential subunits of a complete assembled 19S proteasome complex Six 26S proteasome AAA-ATPase subunits together with four non-ATPase subunits form the base sub complex of 19S regulatory particle for proteasome complex.

KDEL (Lys-Asp-Glu-Leu) endoplasmic reticulum protein retention receptor 1, also known as KDELR1, is a protein which in humans is encoded by the KDELR1 gene.

In molecular biology, protein aggregation is a phenomenon in which intrinsically-disordered or mis-folded proteins aggregate either intra- or extracellularly. Protein aggregates have been implicated in a wide variety of diseases known as amyloidoses, including ALS, Alzheimer's, Parkinson's and prion disease.
Richard I. Morimoto is a Japanese American molecular biologist. He is the Bill and Gayle Cook Professor of Biology and Director of the Rice Institute for Biomedical Research at Northwestern University.
Proteostasis is the dynamic regulation of a balanced, functional proteome. The proteostasis network includes competing and integrated biological pathways within cells that control the biogenesis, folding, trafficking, and degradation of proteins present within and outside the cell. Loss of proteostasis is central to understanding the cause of diseases associated with excessive protein misfolding and degradation leading to loss-of-function phenotypes, as well as aggregation-associated degenerative disorders. Therapeutic restoration of proteostasis may treat or resolve these pathologies.
Chaperone-assisted selective autophagy is a cellular process for the selective, ubiquitin-dependent degradation of chaperone-bound proteins in lysosomes.
Chaperome refers to the ensemble of all cellular molecular chaperone and co-chaperone proteins that assist protein folding of misfolded proteins or folding intermediates in order to ensure native protein folding and function, to antagonize aggregation-related proteotoxicity and ensuing protein loss-of-function or protein misfolding-diseases such as the neurodegenerative diseases Alzheimer's, Huntington's or Parkinson's disease, as well as to safeguard cellular proteostasis and proteome balance.
The mitochondrial unfolded protein response (UPRmt) is a cellular stress response related to the mitochondria. The UPRmt results from unfolded or misfolded proteins in mitochondria beyond the capacity of chaperone proteins to handle them. The UPRmt can occur either in the mitochondrial matrix or in the mitochondrial inner membrane. In the UPRmt, the mitochondrion will either upregulate chaperone proteins or invoke proteases to degrade proteins that fail to fold properly. UPRmt causes the sirtuin SIRT3 to activate antioxidant enzymes and mitophagy.
Anne Bertolotti is a French biochemist and cell biologist who works as Programme Leader at the MRC Laboratory of Molecular Biology in Cambridge, UK. In 2022 she was appointed Head of the MRC LMB's Neurobiology Division. She is known for her research into the cellular defences against misfolded proteins and the mechanisms underlying their deposition, the molecular problem causative of neurodegenerative diseases.
Alfred Lewis Goldberg was an American cell biologist-biochemist and professor at Harvard University. His major discoveries have concerned the mechanisms and physiological importance of protein degradation in cells. Of wide impact have been his lab's demonstration that all cells contain a pathway for selectively eliminating misfolded proteins, his discoveries about the role of proteasomes in this process and of the enzyme systems catalyzing protein breakdown in bacteria, his elucidating the mechanisms for muscle atrophy and the role of proteasomes in antigen presentation to the immune system, and his introduction of proteasome inhibitors now widely used as research tools and in the treatment of blood cancers.



