
Microscopy is the technical field of using microscopes to view objects and areas of objects that cannot be seen with the naked eye. There are three well-known branches of microscopy: optical, electron, and scanning probe microscopy, along with the emerging field of X-ray microscopy.

In molecular biology and biotechnology, a fluorescent tag, also known as a fluorescent label or fluorescent probe, is a molecule that is attached chemically to aid in the detection of a biomolecule such as a protein, antibody, or amino acid. Generally, fluorescent tagging, or labeling, uses a reactive derivative of a fluorescent molecule known as a fluorophore. The fluorophore selectively binds to a specific region or functional group on the target molecule and can be attached chemically or biologically. Various labeling techniques such as enzymatic labeling, protein labeling, and genetic labeling are widely utilized. Ethidium bromide, fluorescein and green fluorescent protein are common tags. The most commonly labelled molecules are antibodies, proteins, amino acids and peptides which are then used as specific probes for detection of a particular target.
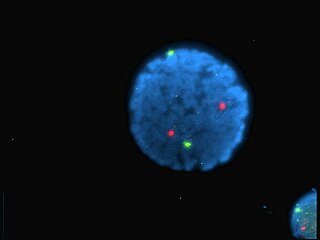
A fluorophore is a fluorescent chemical compound that can re-emit light upon light excitation. Fluorophores typically contain several combined aromatic groups, or planar or cyclic molecules with several π bonds.
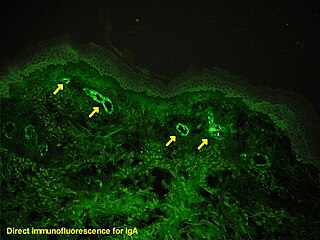
Immunofluorescence is a technique used for light microscopy with a fluorescence microscope and is used primarily on microbiological samples. This technique uses the specificity of antibodies to their antigen to target fluorescent dyes to specific biomolecule targets within a cell, and therefore allows visualization of the distribution of the target molecule through the sample. The specific region an antibody recognizes on an antigen is called an epitope. There have been efforts in epitope mapping since many antibodies can bind the same epitope and levels of binding between antibodies that recognize the same epitope can vary. Additionally, the binding of the fluorophore to the antibody itself cannot interfere with the immunological specificity of the antibody or the binding capacity of its antigen. Immunofluorescence is a widely used example of immunostaining and is a specific example of immunohistochemistry. This technique primarily makes use of fluorophores to visualise the location of the antibodies.

Fluorescence recovery after photobleaching (FRAP) is a method for determining the kinetics of diffusion through tissue or cells. It is capable of quantifying the two dimensional lateral diffusion of a molecularly thin film containing fluorescently labeled probes, or to examine single cells. This technique is very useful in biological studies of cell membrane diffusion and protein binding. In addition, surface deposition of a fluorescing phospholipid bilayer allows the characterization of hydrophilic surfaces in terms of surface structure and free energy.

Förster resonance energy transfer (FRET), fluorescence resonance energy transfer, resonance energy transfer (RET) or electronic energy transfer (EET) is a mechanism describing energy transfer between two light-sensitive molecules (chromophores). A donor chromophore, initially in its electronic excited state, may transfer energy to an acceptor chromophore through nonradiative dipole–dipole coupling. The efficiency of this energy transfer is inversely proportional to the sixth power of the distance between donor and acceptor, making FRET extremely sensitive to small changes in distance.
A total internal reflection fluorescence microscope (TIRFM) is a type of microscope with which a thin region of a specimen, usually less than 200 nanometers can be observed.

A fluorescence microscope is an optical microscope that uses fluorescence instead of, or in addition to, scattering, reflection, and attenuation or absorption, to study the properties of organic or inorganic substances. "Fluorescence microscope" refers to any microscope that uses fluorescence to generate an image, whether it is a simple set up like an epifluorescence microscope or a more complicated design such as a confocal microscope, which uses optical sectioning to get better resolution of the fluorescence image.
Fluorescence correlation spectroscopy (FCS) is a statistical analysis, via time correlation, of stationary fluctuations of the fluorescence intensity. Its theoretical underpinning originated from L. Onsager's regression hypothesis. The analysis provides kinetic parameters of the physical processes underlying the fluctuations. One of the interesting applications of this is an analysis of the concentration fluctuations of fluorescent particles (molecules) in solution. In this application, the fluorescence emitted from a very tiny space in solution containing a small number of fluorescent particles (molecules) is observed. The fluorescence intensity is fluctuating due to Brownian motion of the particles. In other words, the number of the particles in the sub-space defined by the optical system is randomly changing around the average number. The analysis gives the average number of fluorescent particles and average diffusion time, when the particle is passing through the space. Eventually, both the concentration and size of the particle (molecule) are determined. Both parameters are important in biochemical research, biophysics, and chemistry.

In optics, photobleaching is the photochemical alteration of a dye or a fluorophore molecule such that it is permanently unable to fluoresce. This is caused by cleaving of covalent bonds or non-specific reactions between the fluorophore and surrounding molecules. Such irreversible modifications in covalent bonds are caused by transition from a singlet state to the triplet state of the fluorophores. The number of excitation cycles to achieve full bleaching varies. In microscopy, photobleaching may complicate the observation of fluorescent molecules, since they will eventually be destroyed by the light exposure necessary to stimulate them into fluorescing. This is especially problematic in time-lapse microscopy.

Stimulated emission depletion (STED) microscopy is one of the techniques that make up super-resolution microscopy. It creates super-resolution images by the selective deactivation of fluorophores, minimizing the area of illumination at the focal point, and thus enhancing the achievable resolution for a given system. It was developed by Stefan W. Hell and Jan Wichmann in 1994, and was first experimentally demonstrated by Hell and Thomas Klar in 1999. Hell was awarded the Nobel Prize in Chemistry in 2014 for its development. In 1986, V.A. Okhonin had patented the STED idea. This patent was unknown to Hell and Wichmann in 1994.
Sim Scanner is a feature of the Olympus FluoView FV1000 confocal laser scanning microscope. The system incorporates two laser scanners, one for confocal imaging and the other for simultaneous stimulation. They can be illuminated separately and independently, making it possible to stimulate the specimen during observation. As a result, the rapid cell reactions that occur right after laser stimulation can be captured, making the Sim Scanner suitable for such applications as Fluorescence recovery after photobleaching (FRAP), Fluorescence loss in photobleaching (FLIP), photoactivation and photoconversion.
The DyLight Fluor family of fluorescent dyes are produced by Dyomics in collaboration with Thermo Fisher Scientific. DyLight dyes are typically used in biotechnology and research applications as biomolecule, cell and tissue labels for fluorescence microscopy, cell biology or molecular biology.
Lipid bilayer characterization is the use of various optical, chemical and physical probing methods to study the properties of lipid bilayers. Many of these techniques are elaborate and require expensive equipment because the fundamental nature of the lipid bilayer makes it a very difficult structure to study. An individual bilayer, since it is only a few nanometers thick, is invisible in traditional light microscopy. The bilayer is also a relatively fragile structure since it is held together entirely by non-covalent bonds and is irreversibly destroyed if removed from water. In spite of these limitations dozens of techniques have been developed over the last seventy years to allow investigations of the structure and function of bilayers. The first general approach was to utilize non-destructive in situ measurements such as x-ray diffraction and electrical resistance which measured bilayer properties but did not actually image the bilayer. Later, protocols were developed to modify the bilayer and allow its direct visualization at first in the electron microscope and, more recently, with fluorescence microscopy. Over the past two decades, a new generation of characterization tools including AFM has allowed the direct probing and imaging of membranes in situ with little to no chemical or physical modification. More recently, dual polarisation interferometry has been used to measure the optical birefringence of lipid bilayers to characterise order and disruption associated with interactions or environmental effects.

Jennifer Lippincott-Schwartz is a Senior Group Leader at Howard Hughes Medical Institute's Janelia Research Campus and a founding member of the Neuronal Cell Biology Program at Janelia. Previously, she was the Chief of the Section on Organelle Biology in the Cell Biology and Metabolism Program, in the Division of Intramural Research in the Eunice Kennedy Shriver National Institute of Child Health and Human Development at the National Institutes of Health from 1993 to 2016. Lippincott-Schwartz received her PhD from Johns Hopkins University, and performed post-doctoral training with Richard Klausner at the NICHD, NIH in Bethesda, Maryland.

Fluorescence is used in the life sciences generally as a non-destructive way of tracking or analysing biological molecules. Some proteins or small molecules in cells are naturally fluorescent, which is called intrinsic fluorescence or autofluorescence. Alternatively, specific or general proteins, nucleic acids, lipids or small molecules can be "labelled" with an extrinsic fluorophore, a fluorescent dye which can be a small molecule, protein or quantum dot. Several techniques exist to exploit additional properties of fluorophores, such as fluorescence resonance energy transfer, where the energy is passed non-radiatively to a particular neighbouring dye, allowing proximity or protein activation to be detected; another is the change in properties, such as intensity, of certain dyes depending on their environment allowing their use in structural studies.
The FluoProbes series of fluorescent dyes were developed by Interchim to improve performances of standard fluorophores. They are designed for labeling biomolecules, cells, tissues or beads in advanced fluorescent detection techniques.
Super-resolution microscopy is a series of techniques in optical microscopy that allow such images to have resolutions higher than those imposed by the diffraction limit, which is due to the diffraction of light. Super-resolution imaging techniques rely on the near-field or on the far-field. Among techniques that rely on the latter are those that improve the resolution only modestly beyond the diffraction-limit, such as confocal microscopy with closed pinhole or aided by computational methods such as deconvolution or detector-based pixel reassignment, the 4Pi microscope, and structured-illumination microscopy technologies such as SIM and SMI.
Photo-activated localization microscopy and stochastic optical reconstruction microscopy (STORM) are widefield fluorescence microscopy imaging methods that allow obtaining images with a resolution beyond the diffraction limit. The methods were proposed in 2006 in the wake of a general emergence of optical super-resolution microscopy methods, and were featured as Methods of the Year for 2008 by the Nature Methods journal. The development of PALM as a targeted biophysical imaging method was largely prompted by the discovery of new species and the engineering of mutants of fluorescent proteins displaying a controllable photochromism, such as photo-activatible GFP. However, the concomitant development of STORM, sharing the same fundamental principle, originally made use of paired cyanine dyes. One molecule of the pair, when excited near its absorption maximum, serves to reactivate the other molecule to the fluorescent state.
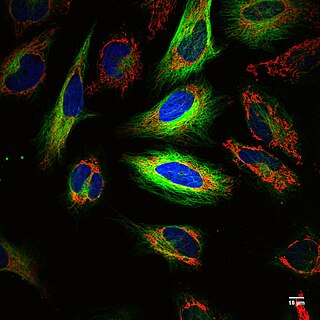
Fluorescence imaging is a type of non-invasive imaging technique that can help visualize biological processes taking place in a living organism. Images can be produced from a variety of methods including: microscopy, imaging probes, and spectroscopy.
