
An alpha helix is a sequence of amino acids in a protein that are twisted into a coil.
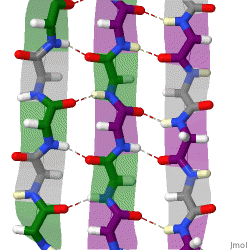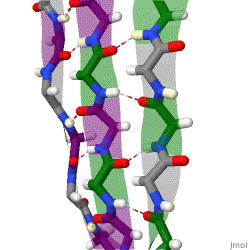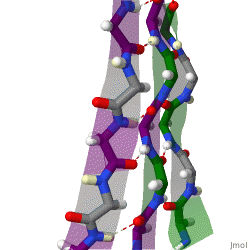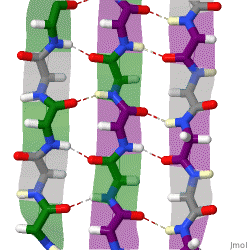
The beta sheet, (β-sheet) is a common motif of the regular protein secondary structure. Beta sheets consist of beta strands (β-strands) connected laterally by at least two or three backbone hydrogen bonds, forming a generally twisted, pleated sheet. A β-strand is a stretch of polypeptide chain typically 3 to 10 amino acids long with backbone in an extended conformation. The supramolecular association of β-sheets has been implicated in the formation of the fibrils and protein aggregates observed in amyloidosis, Alzheimer's disease and other proteinopathies.

Protein secondary structure is the local spatial conformation of the polypeptide backbone excluding the side chains. The two most common secondary structural elements are alpha helices and beta sheets, though beta turns and omega loops occur as well. Secondary structure elements typically spontaneously form as an intermediate before the protein folds into its three dimensional tertiary structure.

Protein folding is the physical process where a protein chain is translated into its native three-dimensional structure, typically a "folded" conformation, by which the protein becomes biologically functional. Via an expeditious and reproducible process, a polypeptide folds into its characteristic three-dimensional structure from a random coil. Each protein exists first as an unfolded polypeptide or random coil after being translated from a sequence of mRNA into a linear chain of amino acids. At this stage, the polypeptide lacks any stable three-dimensional structure. As the polypeptide chain is being synthesized by a ribosome, the linear chain begins to fold into its three-dimensional structure.

Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by computational biology; and it is important in medicine and biotechnology.

Amyloids are aggregates of proteins characterised by a fibrillar morphology of typically 7–13 nm in diameter, a β-sheet secondary structure and ability to be stained by particular dyes, such as Congo red. In the human body, amyloids have been linked to the development of various diseases. Pathogenic amyloids form when previously healthy proteins lose their normal structure and physiological functions (misfolding) and form fibrous deposits within and around cells. These protein misfolding and deposition processes disrupt the healthy function of tissues and organs.

Transthyretin (TTR or TBPA) is a transport protein in the plasma and cerebrospinal fluid that transports the thyroid hormone thyroxine (T4) and retinol to the liver. This is how transthyretin gained its name: transports thyroxine and retinol. The liver secretes TTR into the blood, and the choroid plexus secretes TTR into the cerebrospinal fluid.

Protein structure is the three-dimensional arrangement of atoms in an amino acid-chain molecule. Proteins are polymers – specifically polypeptides – formed from sequences of amino acids, which are the monomers of the polymer. A single amino acid monomer may also be called a residue, which indicates a repeating unit of a polymer. Proteins form by amino acids undergoing condensation reactions, in which the amino acids lose one water molecule per reaction in order to attach to one another with a peptide bond. By convention, a chain under 30 amino acids is often identified as a peptide, rather than a protein. To be able to perform their biological function, proteins fold into one or more specific spatial conformations driven by a number of non-covalent interactions, such as hydrogen bonding, ionic interactions, Van der Waals forces, and hydrophobic packing. To understand the functions of proteins at a molecular level, it is often necessary to determine their three-dimensional structure. This is the topic of the scientific field of structural biology, which employs techniques such as X-ray crystallography, NMR spectroscopy, cryo-electron microscopy (cryo-EM) and dual polarisation interferometry, to determine the structure of proteins.
Site-directed spin labeling (SDSL) is a technique for investigating the structure and local dynamics of proteins using electron spin resonance. The theory of SDSL is based on the specific reaction of spin labels with amino acids. A spin label's built-in protein structure can be detected by EPR spectroscopy. SDSL is also a useful tool in examinations of the protein folding process.

Amylin, or islet amyloid polypeptide (IAPP), is a 37-residue peptide hormone. It is co-secreted with insulin from the pancreatic β-cells in the ratio of approximately 100:1 (insulin:amylin). Amylin plays a role in glycemic regulation by slowing gastric emptying and promoting satiety, thereby preventing post-prandial spikes in blood glucose levels.

A fungal prion is a prion that infects hosts which are fungi. Fungal prions are naturally occurring proteins that can switch between multiple, structurally distinct conformations, at least one of which is self-propagating and transmissible to other prions. This transmission of protein state represents an epigenetic phenomenon where information is encoded in the protein structure itself, instead of in nucleic acids. Several prion-forming proteins have been identified in fungi, primarily in the yeast Saccharomyces cerevisiae. These fungal prions are generally considered benign, and in some cases even confer a selectable advantage to the organism.
A beta bulge can be described as a localized disruption of the regular hydrogen bonding of beta sheet by inserting extra residues into one or both hydrogen bonded β-strands.

A 310 helix is a type of secondary structure found in proteins and polypeptides. Of the numerous protein secondary structures present, the 310-helix is the fourth most common type observed; following α-helices, β-sheets and reverse turns. 310-helices constitute nearly 10–15% of all helices in protein secondary structures, and are typically observed as extensions of α-helices found at either their N- or C- termini. Because of the α-helices tendency to consistently fold and unfold, it has been proposed that the 310-helix serves as an intermediary conformation of sorts, and provides insight into the initiation of α-helix folding.
Peptide plane flipping is a type of conformational change that can occur in proteins by which the dihedral angles of adjacent amino acids undergo large-scale rotations with little displacement of the side chains. The plane flip is defined as a rotation of the dihedral angles φ,ψ at amino acids i and i+1 such that the resulting angles remain in structurally stable regions of Ramachandran space. The key requirement is that the sum of the ψi angle of residue i and the φi+1 angle of residue i+1 remain roughly constant; in effect, the flip is a crankshaft move about the axis defined by the Cα-C¹ and N-Cα bond vectors of the peptide group, which are roughly parallel. As an example, the type I and type II beta turns differ by a simple flip of the central peptide group of the turn.

Collagen alpha-1(XXV) chain is a protein that in humans is encoded by the COL25A1 gene.

In molecular biology, the GCM transcription factors are a family of proteins which contain a GCM motif. The GCM motif is a domain that has been identified in proteins belonging to a family of transcriptional regulators involved in fundamental developmental processes which comprise Drosophila melanogaster GCM and its mammalian homologues. In GCM transcription factors the N-terminal moiety contains a DNA-binding domain of 150 amino acids. Sequence conservation is highest in this GCM domain. In contrast, the C-terminal moiety contains one or two transactivating regions and is only poorly conserved.
The Nest is a type of protein structural motif. It is a small recurring anion-binding feature of both proteins and peptides. Each consists of the main chain atoms of three consecutive amino acid residues. The main chain NH groups bind the anions while the side chain atoms are often not involved. Proline residues lack NH groups so are rare in nests. About one in 12 of amino acid residues in proteins, on average, belongs to a nest.

p3 peptide also known as amyloid β- peptide (Aβ)17–40/42 is the peptide resulting from the α- and γ-secretase cleavage from the amyloid precursor protein (APP). It is known to be the major constituent of diffuse plaques observed in Alzheimer's disease (AD) brains and pre-amyloid plaques in people affected by Down syndrome. However, p3 peptide's role in these diseases is not truly known yet.
The beta bend ribbon, or beta-bend ribbon, is a structural feature in polypeptides and proteins. The shortest possible has six amino acid residues arranged as two overlapping hydrogen bonded beta turns in which the carbonyl group of residue i is hydrogen-bonded to the NH of residue i+3 while the carbonyl group of residue i+2 is hydrogen-bonded to the NH of residue i+5. In longer ribbons, this bonding is continued in peptides of 8, 10, etc., amino acid residues. A beta bend ribbon can be regarded as an aberrant 310 helix (3/10-helix) that has lost some of its hydrogen bonds. Two websites are available to facilitate finding and examining these features in proteins: Motivated Proteins; and PDBeMotif.
Computational methods that use protein sequence and/ or protein structure to predict protein aggregation. The table below, shows the main features of software for prediction of protein aggregation

