Amyloids are aggregates of proteins characterised by a fibrillar morphology of typically 7–13 nm in diameter, a β-sheet secondary structure and ability to be stained by particular dyes, such as Congo red. In the human body, amyloids have been linked to the development of various diseases. Pathogenic amyloids form when previously healthy proteins lose their normal structure and physiological functions (misfolding) and form fibrous deposits within and around cells. These protein misfolding and deposition processes disrupt the healthy function of tissues and organs.

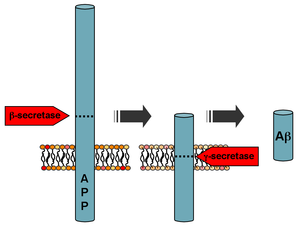
Amyloid-beta precursor protein (APP) is an integral membrane protein expressed in many tissues and concentrated in the synapses of neurons. It functions as a cell surface receptor and has been implicated as a regulator of synapse formation, neural plasticity, antimicrobial activity, and iron export. It is coded for by the gene APP and regulated by substrate presentation. APP is best known as the precursor molecule whose proteolysis generates amyloid beta (Aβ), a polypeptide containing 37 to 49 amino acid residues, whose amyloid fibrillar form is the primary component of amyloid plaques found in the brains of Alzheimer's disease patients.

Amyloid plaques are extracellular deposits of amyloid beta (Aβ) protein that present mainly in the grey matter of the brain. Degenerative neuronal elements and an abundance of microglia and astrocytes can be associated with amyloid plaques. Some plaques occur in the brain as a result of aging, but large numbers of plaques and neurofibrillary tangles are characteristic features of Alzheimer's disease. The plaques are highly variable in shape and size; in tissue sections immunostained for Aβ, they comprise a log-normal size distribution curve, with an average plaque area of 400-450 square micrometers (μm2). The smallest plaques, which often consist of diffuse deposits of Aβ, are particularly numerous. Plaques form when Aβ misfolds and aggregates into oligomers and longer polymers, the latter of which are characteristic of amyloid.
Pittsburgh compound B (PiB) is a radioactive analog of thioflavin T, which can be used in positron emission tomography scans to image beta-amyloid plaques in neuronal tissue. Due to this property, Pittsburgh compound B may be used in investigational studies of Alzheimer's disease.

Thioflavins are fluorescent dyes that are available as at least two compounds, namely Thioflavin T and Thioflavin S. Both are used for histology staining and biophysical studies of protein aggregation. In particular, these dyes have been used since 1989 to investigate amyloid formation. They are also used in biophysical studies of the electrophysiology of bacteria. Thioflavins are corrosive, irritant, and acutely toxic, causing serious eye damage. Thioflavin T has been used in research into Alzheimer's disease and other neurodegenerative diseases.
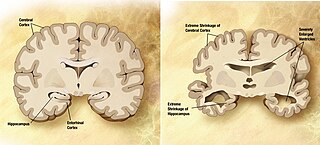
A neurodegenerative disease is caused by the progressive loss of neurons, in the process known as neurodegeneration. Neuronal damage may also ultimately result in their death. Neurodegenerative diseases include amyotrophic lateral sclerosis, multiple sclerosis, Parkinson's disease, Alzheimer's disease, Huntington's disease, multiple system atrophy, tauopathies, and prion diseases. Neurodegeneration can be found in the brain at many different levels of neuronal circuitry, ranging from molecular to systemic. Because there is no known way to reverse the progressive degeneration of neurons, these diseases are considered to be incurable; however research has shown that the two major contributing factors to neurodegeneration are oxidative stress and inflammation. Biomedical research has revealed many similarities between these diseases at the subcellular level, including atypical protein assemblies and induced cell death. These similarities suggest that therapeutic advances against one neurodegenerative disease might ameliorate other diseases as well.

Beta-secretase 1, also known as beta-site amyloid precursor protein cleaving enzyme 1, beta-site APP cleaving enzyme 1 (BACE1), membrane-associated aspartic protease 2, memapsin-2, aspartyl protease 2, and ASP2, is an enzyme that in humans is encoded by the BACE1 gene. Expression of BACE1 is observed mainly in neurons and oligodendrocytes.
The biochemistry of Alzheimer's disease, the most common cause of dementia, is not yet very well understood. Alzheimer's disease (AD) has been identified as a proteopathy: a protein misfolding disease due to the accumulation of abnormally folded amyloid beta (Aβ) protein in the brain. Amyloid beta is a short peptide that is an abnormal proteolytic byproduct of the transmembrane protein amyloid-beta precursor protein (APP), whose function is unclear but thought to be involved in neuronal development. The presenilins are components of proteolytic complex involved in APP processing and degradation.

Presenilins are a family of related multi-pass transmembrane proteins which constitute the catalytic subunits of the gamma-secretase intramembrane protease protein complex. They were first identified in screens for mutations causing early onset forms of familial Alzheimer's disease by Peter St George-Hyslop. Vertebrates have two presenilin genes, called PSEN1 that codes for presenilin 1 (PS-1) and PSEN2 that codes for presenilin 2 (PS-2). Both genes show conservation between species, with little difference between rat and human presenilins. The nematode worm C. elegans has two genes that resemble the presenilins and appear to be functionally similar, sel-12 and hop-1.
APH-1 is a protein originally identified in the round worm Caenorhabditis elegans as a regulator of the cell-surface localization of nicastrin in the Notch signaling pathway.

In medicine, proteinopathy, or proteopathy, protein conformational disorder, or protein misfolding disease, is a class of diseases in which certain proteins become structurally abnormal, and thereby disrupt the function of cells, tissues and organs of the body.

Insulin-degrading enzyme, also known as IDE, is an enzyme.

Collagen alpha-1(XXV) chain is a protein that in humans is encoded by the COL25A1 gene.
Early-onset Alzheimer's disease (EOAD), also called younger-onset Alzheimer's disease (YOAD), is Alzheimer's disease diagnosed before the age of 65. It is an uncommon form of Alzheimer's, accounting for only 5–10% of all Alzheimer's cases. About 60% have a positive family history of Alzheimer's and 13% of them are inherited in an autosomal dominant manner. Most cases of early-onset Alzheimer's share the same traits as the "late-onset" form and are not caused by known genetic mutations. Little is understood about how it starts.
Solanezumab is a monoclonal antibody being investigated by Eli Lilly as a neuroprotector for patients with Alzheimer's disease. The drug originally attracted extensive media coverage proclaiming it a breakthrough, but it has failed to show promise in Phase III trials.
The biomarkers of Alzheimer's disease are neurochemical indicators used to assess the risk or presence of the disease. The biomarkers can be used to diagnose Alzheimer's disease (AD) in a very early stage, but they also provide objective and reliable measures of disease progress. It is imperative to diagnose AD disease as soon as possible, because neuropathologic changes of AD precede the symptoms by years. It is well known that amyloid beta (Aβ) is a good indicator of AD disease, which has facilitated doctors to accurately pre-diagnose cases of AD. When Aβ peptide is released by proteolytic cleavage of amyloid-beta precursor protein, some Aβ peptides that are solubilized are detected in CSF and blood plasma which makes AB peptides a promising candidate for biological markers. It has been shown that the amyloid beta biomarker shows 80% or above sensitivity and specificity, in distinguishing AD from dementia. It is believed that amyloid beta as a biomarker will provide a future for diagnosis of AD and eventually treatment of AD.

p3 peptide also known as amyloid β- peptide (Aβ)17–40/42 is the peptide resulting from the α- and γ-secretase cleavage from the amyloid precursor protein (APP). It is known to be the major constituent of diffuse plaques observed in Alzheimer's disease (AD) brains and pre-amyloid plaques in people affected by Down syndrome. However, p3 peptide's role in these diseases is not truly known yet.
The ion channel hypothesis of Alzheimer's disease (AD), also known as the channel hypothesis or the amyloid beta ion channel hypothesis, is a more recent variant of the amyloid hypothesis of AD, which identifies amyloid beta (Aβ) as the underlying cause of neurotoxicity seen in AD. While the traditional formulation of the amyloid hypothesis pinpoints insoluble, fibrillar aggregates of Aβ as the basis of disruption of calcium ion homeostasis and subsequent apoptosis in AD, the ion channel hypothesis in 1993 introduced the possibility of an ion-channel-forming oligomer of soluble, non-fibrillar Aβ as the cytotoxic species allowing unregulated calcium influx into neurons in AD.
Dennis J. Selkoe is an American physician (neurologist) known for his research into the molecular basis of Alzheimer's disease. In 1985 he became Co-Director of the Center for Neurological Diseases and from 1990, Vincent and Stella Coates Professor of Neurological Diseases at Harvard Medical School. He is also a Fellow of the AAAS and a member of the National Academy of Medicine.

Buntanetap is an orally-administered small molecule inhibitor of several neurotoxic proteins that is under investigation in the treatment of Alzheimer's disease, frontotemporal dementia, chronic traumatic encephalopathy and Parkinson's disease. It is the (+) enantiomer of phenserine, as the (-) enantiomer also has unwanted anticholinergic effects. It is currently in phase III trials for the treatment of Parkinson's.