






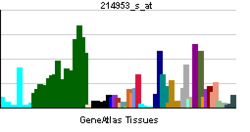

Amyloid-beta precursor protein (APP) is an integral membrane protein expressed in many tissues and concentrated in the synapses of neurons. It functions as a cell surface receptor [5] and has been implicated as a regulator of synapse formation, [6] neural plasticity, [7] antimicrobial activity, [8] and iron export. [9] It is coded for by the gene APP and regulated by substrate presentation. [10] APP is best known as the precursor molecule whose proteolysis generates amyloid beta (Aβ), a polypeptide containing 37 to 49 amino acid residues, whose amyloid fibrillar form is the primary component of amyloid plaques found in the brains of Alzheimer's disease patients.


























