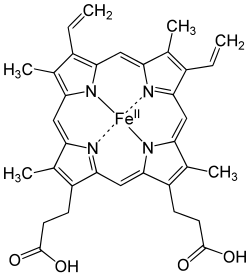
Dietary iron uptake
The absorption of dietary iron is a variable and dynamic process. The amount of iron absorbed compared to the amount ingested is typically low, but may range from 5% to as much as 35% depending on circumstances and type of iron. The efficiency with which iron is absorbed varies depending on the source. Generally, the best-absorbed forms of iron come from animal products. Absorption of dietary iron in iron salt form (as in most supplements) varies somewhat according to the body's need for iron, and is usually between 10% and 20% of iron intake. Absorption of iron from animal products, and some plant products, is in the form of heme iron, and is more efficient, allowing absorption of from 15% to 35% of intake. Heme iron in animals is from blood and heme-containing proteins in meat and mitochondria, whereas in plants, heme iron is present in mitochondria in all cells that use oxygen for respiration.
Like most mineral nutrients, the majority of the iron absorbed from digested food or supplements is absorbed in the duodenum by enterocytes of the duodenal lining. These cells have special molecules that allow them to move iron into the body. To be absorbed, dietary iron can be absorbed as part of a protein such as heme protein or iron must be in its ferrous Fe2+ form. A ferric reductase enzyme on the enterocytes' brush border, duodenal cytochrome B (Dcytb), reduces ferric Fe3+ to Fe2+. [11] A protein called divalent metal transporter 1 (DMT1), which can transport several divalent metals across the plasma membrane, then transports iron across the enterocyte's cell membrane into the cell. If the iron is bound to heme, it is instead transported across the apical membrane by heme carrier protein 1 (HCP1). [12] Heme is then catabolized by microsomal heme oxygenase into biliverdin, releasing Fe2+. [13]
These intestinal lining cells can then either store the iron as ferritin, which is accomplished by Fe2+ binding to apoferritin (in which case the iron will leave the body when the cell dies and is sloughed off into feces), or the cell can release it into the body via the only known iron exporter in mammals, ferroportin. Hephaestin, a ferroxidase that can oxidize Fe2+ to Fe3+ and is found mainly in the small intestine, helps ferroportin transfer iron across the basolateral end of the intestine cells. Upon release into the bloodstream, Fe3+ binds transferrin and circulates to tissues. In contrast, ferroportin is post-translationally repressed by hepcidin, a 25-amino acid peptide hormone. The body regulates iron levels by regulating each of these steps. For instance, enterocytes synthesize more Dcytb, DMT1 and ferroportin in response to iron deficiency anemia. [14] Iron absorption from diet is enhanced in the presence of vitamin C and diminished by excess calcium, zinc, or manganese. [15]
The human body's rate of iron absorption appears to respond to a variety of interdependent factors, including total iron stores, the extent to which the bone marrow is producing new red blood cells, the concentration of hemoglobin in the blood, and the oxygen content of the blood. The body also absorbs less iron during times of inflammation, in order to deprive bacteria of iron. Recent discoveries demonstrate that hepcidin regulation of ferroportin is responsible for the syndrome of anemia of chronic disease.
Iron recycling and loss
Most of the iron in the body is hoarded and recycled by the reticuloendothelial system, which breaks down aged red blood cells. In contrast to iron uptake and recycling, there is no physiologic regulatory mechanism for excreting iron. People lose a small but steady amount by gastrointestinal blood loss, sweating and by shedding cells of the skin and the mucosal lining of the gastrointestinal tract. The total amount of loss for healthy people in the developed world amounts to an estimated average of 1 mg a day for men, and 1.5–2 mg a day for women with regular menstrual periods. [16] People with gastrointestinal parasitic infections, more commonly found in developing countries, often lose more. [2] Those who cannot regulate absorption well enough get disorders of iron overload. In these diseases, the toxicity of iron starts overwhelming the body's ability to bind and store it. [17]