
The alpha helix (α-helix) is a common motif in the secondary structure of proteins and is a right hand-helix conformation in which every backbone N−H group hydrogen bonds to the backbone C=O group of the amino acid located four residues earlier along the protein sequence.

The beta sheet, (β-sheet) is a common motif of the regular protein secondary structure. Beta sheets consist of beta strands (β-strands) connected laterally by at least two or three backbone hydrogen bonds, forming a generally twisted, pleated sheet. A β-strand is a stretch of polypeptide chain typically 3 to 10 amino acids long with backbone in an extended conformation. The supramolecular association of β-sheets has been implicated in the formation of the fibrils and protein aggregates observed in amyloidosis, notably Alzheimer's disease.

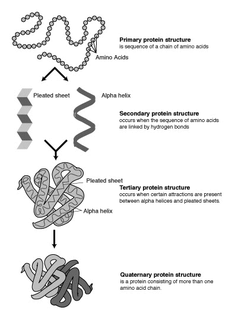
Protein secondary structure is the three dimensional form of local segments of proteins. The two most common secondary structural elements are alpha helices and beta sheets, though beta turns and omega loops occur as well. Secondary structure elements typically spontaneously form as an intermediate before the protein folds into its three dimensional tertiary structure.

Proline (symbol Pro or P) is an organic acid classed as a proteinogenic amino acid (used in the biosynthesis of proteins), although it does not contain the amino group -NH
2 but is rather a secondary amine. The secondary amine nitrogen is in the protonated NH2+ form under biological conditions, while the carboxy group is in the deprotonated −COO− form. The "side chain" from the α carbon connects to the nitrogen forming a pyrrolidine loop, classifying it as a aliphatic amino acid. It is non-essential in humans, meaning the body can synthesize it from the non-essential amino acid L-glutamate. It is encoded by all the codons starting with CC (CCU, CCC, CCA, and CCG).
Protein structure prediction is the inference of the three-dimensional structure of a protein from its amino acid sequence—that is, the prediction of its secondary and tertiary structure from primary structure. Structure prediction is different from the inverse problem of protein design. Protein structure prediction is one of the most important goals pursued by computational biology; and it is important in medicine and biotechnology.
In a chain-like biological molecule, such as a protein or nucleic acid, a structural motif is a common three-dimensional structure that appears in a variety of different, evolutionarily unrelated molecules. A structural motif does not have to be associated with a sequence motif; it can be represented by different and completely unrelated sequences in different proteins or RNA.

Protein structure is the three-dimensional arrangement of atoms in an amino acid-chain molecule. Proteins are polymers – specifically polypeptides – formed from sequences of amino acids, the monomers of the polymer. A single amino acid monomer may also be called a residue indicating a repeating unit of a polymer. Proteins form by amino acids undergoing condensation reactions, in which the amino acids lose one water molecule per reaction in order to attach to one another with a peptide bond. By convention, a chain under 30 amino acids is often identified as a peptide, rather than a protein. To be able to perform their biological function, proteins fold into one or more specific spatial conformations driven by a number of non-covalent interactions such as hydrogen bonding, ionic interactions, Van der Waals forces, and hydrophobic packing. To understand the functions of proteins at a molecular level, it is often necessary to determine their three-dimensional structure. This is the topic of the scientific field of structural biology, which employs techniques such as X-ray crystallography, NMR spectroscopy, cryo electron microscopy (cryo-EM) and dual polarisation interferometry to determine the structure of proteins.
A supersecondary structure is a compact three-dimensional protein structure of several adjacent elements of a secondary structure that is smaller than a protein domain or a subunit. Supersecondary structures can act as nucleations in the process of protein folding.

In protein structures, a beta barrel is a beta-sheet composed of tandem repeats that twists and coils to form a closed toroidal structure in which the first strand is bonded to the last strand. Beta-strands in many beta-barrels are arranged in an antiparallel fashion. Beta barrel structures are named for resemblance to the barrels used to contain liquids. Most of them are water-soluble proteins and frequently bind hydrophobic ligands in the barrel center, as in lipocalins. Others span cell membranes and commonly found in porins. Porin-like barrel structures are encoded by as many as 2–3% of the genes in Gram-negative bacteria.
A turn is an element of secondary structure in proteins where the polypeptide chain reverses its overall direction.
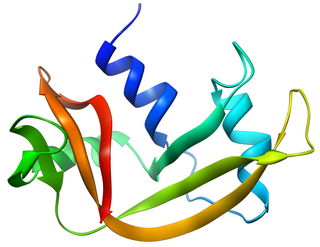
Bovine pancreatic ribonuclease, also often referred to as bovine pancreatic ribonuclease A or simply RNase A, is a pancreatic ribonuclease enzyme that cleaves single-stranded RNA. Bovine pancreatic ribonuclease is one of the classic model systems of protein science. Two Nobel Prizes in Chemistry have been awarded in recognition of work on bovine pancreatic ribonuclease: in 1972, the Prize was awarded to Christian Anfinsen for his work on protein folding and to Stanford Moore and William Stein for their work on the relationship between the protein's structure and its chemical mechanism; in 1984, the Prize was awarded to Robert Bruce Merrifield for development of chemical synthesis of proteins.
A beta bulge can be described as a localized disruption of the regular hydrogen bonding of beta sheet by inserting extra residues into one or both hydrogen bonded β-strands.
A polyproline helix is a type of protein secondary structure which occurs in proteins comprising repeating proline residues. A left-handed polyproline II helix is formed when sequential residues all adopt (φ,ψ) backbone dihedral angles of roughly and have trans isomers of their peptide bonds. This PPII conformation is also common in proteins and polypeptides with other amino acids apart from proline. Similarly, a more compact right-handed polyproline I helix is formed when sequential residues all adopt (φ,ψ) backbone dihedral angles of roughly and have cis isomers of their peptide bonds. Of the twenty common naturally occurring amino acids, only proline is likely to adopt the cis isomer of the peptide bond, specifically the X-Pro peptide bond; steric and electronic factors heavily favor the trans isomer in most other peptide bonds. However, peptide bonds that replace proline with another N-substituted amino acid are also likely to adopt the cis isomer.

Prolyl isomerase is an enzyme found in both prokaryotes and eukaryotes that interconverts the cis and trans isomers of peptide bonds with the amino acid proline. Proline has an unusually conformationally restrained peptide bond due to its cyclic structure with its side chain bonded to its secondary amine nitrogen. Most amino acids have a strong energetic preference for the trans peptide bond conformation due to steric hindrance, but proline's unusual structure stabilizes the cis form so that both isomers are populated under biologically relevant conditions. Proteins with prolyl isomerase activity include cyclophilin, FKBPs, and parvulin, although larger proteins can also contain prolyl isomerase domains.

A leucine-rich repeat (LRR) is a protein structural motif that forms an α/β horseshoe fold. It is composed of repeating 20–30 amino acid stretches that are unusually rich in the hydrophobic amino acid leucine. These tandem repeats commonly fold together to form a solenoid protein domain, termed leucine-rich repeat domain. Typically, each repeat unit has beta strand-turn-alpha helix structure, and the assembled domain, composed of many such repeats, has a horseshoe shape with an interior parallel beta sheet and an exterior array of helices. One face of the beta sheet and one side of the helix array are exposed to solvent and are therefore dominated by hydrophilic residues. The region between the helices and sheets is the protein's hydrophobic core and is tightly sterically packed with leucine residues.
Protegrins are small peptides containing 16-18 amino acid residues. Protegrins were first discovered in porcine leukocytes and were found to have antimicrobial activity against bacteria, fungi, and some enveloped viruses. The amino acid composition of protegrins contains six positively charged arginine residues and four cysteine residues. Their secondary structure is classified as cysteine-rich β-sheet antimicrobial peptides, AMPs, that display limited sequence similarity to certain defensins and tachyplesins. In solution, the peptides fold to form an anti-parallel β-strand with the structure stabilized by two cysteine bridges formed among the four cysteine residues. Recent studies suggest that protegrins can bind to lipopolysaccharide, a property that may help them to insert into the membranes of gram-negative bacteria and permeabilize them.
β turns are the most common form of turns—a type of non-regular secondary structure in proteins that cause a change in direction of the polypeptide chain. They are very common motifs in proteins and polypeptides. Each consists of four amino acid residues. They can be defined in two ways: 1. By the possession of an intra-main-chain hydrogen bond between the CO of residue i and the NH of residue i+3; Alternatively, 2. By having a distance of less than 7Å between the Cα atoms of residues i and i+3. The hydrogen bond criterion is the one most appropriate for everyday use, partly because it gives rise to four distinct categories; the distance criterion gives rise to the same four categories but yields additional turn types.
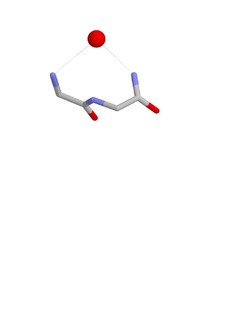
The Nest is a type of protein structural motif. It is a small recurring anion-binding feature of both proteins and peptides. Each consists of the main chain atoms of three consecutive amino acid residues. The main chain NH groups bind the anions while the side chain atoms are often not involved. Proline residues lack NH groups so are rare in nests. About one in 12 of amino acid residues in proteins, on average, belongs to a nest.

Schellman loops are commonly occurring structural features of proteins and polypeptides. Each has six amino acid residues with two specific inter-mainchain hydrogen bonds and a characteristic main chain dihedral angle conformation. The CO group of residue i is hydrogen-bonded to the NH of residue i+5, and the CO group of residue i+1 is hydrogen-bonded to the NH of residue i+4. Residues i+1, i+2, and i+3 have negative φ (phi) angle values and the phi value of residue i+4 is positive. Schellman loops incorporate a three amino acid residue RL nest, in which three mainchain NH groups form a concavity for hydrogen bonding to carbonyl oxygens. About 2.5% of amino acids in proteins belong to Schellman loops. Two websites are available for examining small motifs in proteins, Motivated Proteins: ; or PDBeMotif:.

Beta bulge loops are commonly occurring motifs in proteins and polypeptides consisting of five to six amino acids. There are two types: type 1, which is a pentapeptide; and type 2, with six amino acids. They are regarded as a type of beta bulge, and have the alternative name of type G1 beta bulge. Compared to other beta bulges, beta bulge loops give rise to chain reversal such that they often occur at the loop ends of beta hairpins; hairpins of this sort can be described as 3:5 or 4:6. Two websites are available for finding and examining β bulge loops in proteins, Motivated Proteins: and PDBeMotif:.





