A chemical database is a database specifically designed to store chemical information. This information is about chemical and crystal structures, spectra, reactions and syntheses, and thermophysical data.
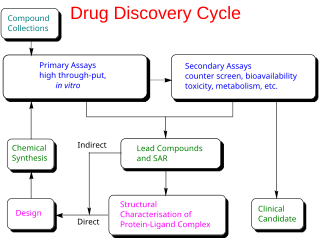
In the fields of medicine, biotechnology, and pharmacology, drug discovery is the process by which new candidate medications are discovered.
Cheminformatics refers to the use of physical chemistry theory with computer and information science techniques—so called "in silico" techniques—in application to a range of descriptive and prescriptive problems in the field of chemistry, including in its applications to biology and related molecular fields. Such in silico techniques are used, for example, by pharmaceutical companies and in academic settings to aid and inform the process of drug discovery, for instance in the design of well-defined combinatorial libraries of synthetic compounds, or to assist in structure-based drug design. The methods can also be used in chemical and allied industries, and such fields as environmental science and pharmacology, where chemical processes are involved or studied.
In the physical sciences, a partition coefficient (P) or distribution coefficient (D) is the ratio of concentrations of a compound in a mixture of two immiscible solvents at equilibrium. This ratio is therefore a comparison of the solubilities of the solute in these two liquids. The partition coefficient generally refers to the concentration ratio of un-ionized species of compound, whereas the distribution coefficient refers to the concentration ratio of all species of the compound.

Medicinal or pharmaceutical chemistry is a scientific discipline at the intersection of chemistry and pharmacy involved with designing and developing pharmaceutical drugs. Medicinal chemistry involves the identification, synthesis and development of new chemical entities suitable for therapeutic use. It also includes the study of existing drugs, their biological properties, and their quantitative structure-activity relationships (QSAR).

In medicinal chemistry and molecular biology, a pharmacophore is an abstract description of molecular features that are necessary for molecular recognition of a ligand by a biological macromolecule. IUPAC defines a pharmacophore to be "an ensemble of steric and electronic features that is necessary to ensure the optimal supramolecular interactions with a specific biological target and to trigger its biological response". A pharmacophore model explains how structurally diverse ligands can bind to a common receptor site. Furthermore, pharmacophore models can be used to identify through de novo design or virtual screening novel ligands that will bind to the same receptor.
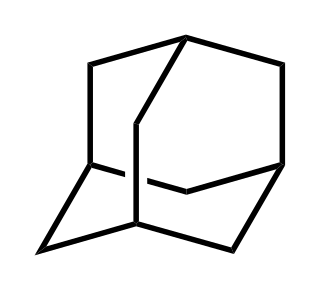
Adamantane is an organic compound with formula C10H16 or, more descriptively, (CH)4(CH2)6. Adamantane molecules can be described as the fusion of three cyclohexane rings. The molecule is both rigid and virtually stress-free. Adamantane is the most stable isomer of C10H16. The spatial arrangement of carbon atoms in the adamantane molecule is the same as in the diamond crystal. This similarity led to the name adamantane, which is derived from the Greek adamantinos (relating to steel or diamond). It is a white solid with a camphor-like odor. It is the simplest diamondoid.
Chemical space is a concept in cheminformatics referring to the property space spanned by all possible molecules and chemical compounds adhering to a given set of construction principles and boundary conditions. It contains millions of compounds which are readily accessible and available to researchers. It is a library used in the method of molecular docking.

In the field of molecular modeling, docking is a method which predicts the preferred orientation of one molecule to a second when a ligand and a target are bound to each other to form a stable complex. Knowledge of the preferred orientation in turn may be used to predict the strength of association or binding affinity between two molecules using, for example, scoring functions.
A photoswitch is a type of molecule that can change its structural geometry and chemical properties upon irradiation with electromagnetic radiation. Although often used interchangeably with the term molecular machine, a switch does not perform work upon a change in its shape whereas a machine does. However, photochromic compounds are the necessary building blocks for light driven molecular motors and machines. Upon irradiation with light, photoisomerization about double bonds in the molecule can lead to changes in the cis- or trans- configuration. These photochromic molecules are being considered for a range of applications.
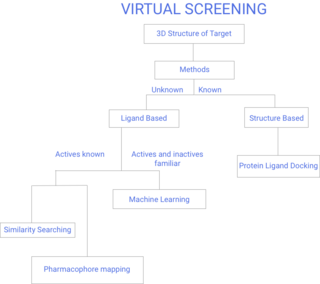
Virtual screening (VS) is a computational technique used in drug discovery to search libraries of small molecules in order to identify those structures which are most likely to bind to a drug target, typically a protein receptor or enzyme.
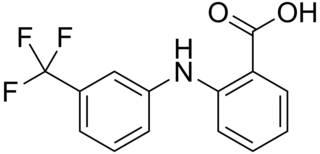
Flufenamic acid (FFA) is a member of the anthranilic acid derivatives class of nonsteroidal anti-inflammatory drugs (NSAIDs). Like other members of the class, it is a cyclooxygenase (COX) inhibitor, preventing the formation of prostaglandins. FFA is known to bind to and reduce the activity of prostaglandin F synthase and activate TRPC6.
In the fields of chemical graph theory, molecular topology, and mathematical chemistry, a topological index, also known as a connectivity index, is a type of a molecular descriptor that is calculated based on the molecular graph of a chemical compound. Topological indices are numerical parameters of a graph which characterize its topology and are usually graph invariant. Topological indices are used for example in the development of quantitative structure-activity relationships (QSARs) in which the biological activity or other properties of molecules are correlated with their chemical structure.

Chemical similarity refers to the similarity of chemical elements, molecules or chemical compounds with respect to either structural or functional qualities, i.e. the effect that the chemical compound has on reaction partners in inorganic or biological settings. Biological effects and thus also similarity of effects are usually quantified using the biological activity of a compound. In general terms, function can be related to the chemical activity of compounds.
Inte:Ligand was founded in Maria Enzersdorf, Lower Austria (Niederösterreich) in 2003. They established the company headquarters on Mariahilferstrasse in Vienna, Austria that same year.
Fragment-based lead discovery (FBLD) also known as fragment-based drug discovery (FBDD) is a method used for finding lead compounds as part of the drug discovery process. Fragments are small organic molecules which are small in size and low in molecular weight. It is based on identifying small chemical fragments, which may bind only weakly to the biological target, and then growing them or combining them to produce a lead with a higher affinity. FBLD can be compared with high-throughput screening (HTS). In HTS, libraries with up to millions of compounds, with molecular weights of around 500 Da, are screened, and nanomolar binding affinities are sought. In contrast, in the early phase of FBLD, libraries with a few thousand compounds with molecular weights of around 200 Da may be screened, and millimolar affinities can be considered useful. FBLD is a technique being used in research for discovering novel potent inhibitors. This methodology could help to design multitarget drugs for multiple diseases. The multitarget inhibitor approach is based on designing an inhibitor for the multiple targets. This type of drug design opens up new polypharmacological avenues for discovering innovative and effective therapies. Neurodegenerative diseases like Alzheimer’s (AD) and Parkinson’s, among others, also show rather complex etiopathologies. Multitarget inhibitors are more appropriate for addressing the complexity of AD and may provide new drugs for controlling the multifactorial nature of AD, stopping its progression.
Topological inhibitors are rigid three-dimensional molecules of inorganic, organic, and hybrid compounds that form multicentered supramolecular interactions in vacant cavities of protein macromolecules and their complexes.
Matched molecular pair analysis (MMPA) is a method in cheminformatics that compares the properties of two molecules that differ only by a single chemical transformation, such as the substitution of a hydrogen atom by a chlorine one. Such pairs of compounds are known as matched molecular pairs (MMP). Because the structural difference between the two molecules is small, any experimentally observed change in a physical or biological property between the matched molecular pair can more easily be interpreted. The term was first coined by Kenny and Sadowski in the book Chemoinformatics in Drug Discovery.
Igor Volodymyrovych Komarov is a Ukrainian synthetic organic chemist, specializing in medicinal chemistry and nanotechnology. He is the director of the Institute of High Technologies of Taras Shevchenko National University of Kyiv. He is also a scientific advisor of Enamine Ltd (Ukraine) and Lumobiotics GmbH (Germany).

Iwao Ojima is a Japanese-American chemist and university distinguished professor at the State University of New York at Stony Brook. He has been widely recognized for his seminal contributions to a range of chemical research at the multifaceted interfaces of chemical synthesis and life sciences. As rare accomplishments, he has received four National Awards from the American Chemical Society in four different fields of research. He is also serving as the director of the Institute of Chemical Biology and Drug Discovery (ICB&DD), as well as the president of the Stony Brook Chapter of the National Academy of Inventors.

