Related Research Articles

Molecular diffusion, often simply called diffusion, is the thermal motion of all particles at temperatures above absolute zero. The rate of this movement is a function of temperature, viscosity of the fluid and the size (mass) of the particles. Diffusion explains the net flux of molecules from a region of higher concentration to one of lower concentration. Once the concentrations are equal the molecules continue to move, but since there is no concentration gradient the process of molecular diffusion has ceased and is instead governed by the process of self-diffusion, originating from the random motion of the molecules. The result of diffusion is a gradual mixing of material such that the distribution of molecules is uniform. Since the molecules are still in motion, but an equilibrium has been established, the result of molecular diffusion is called a "dynamic equilibrium". In a phase with uniform temperature, absent external net forces acting on the particles, the diffusion process will eventually result in complete mixing.

Physical chemistry is the study of macroscopic and microscopic phenomena in chemical systems in terms of the principles, practices, and concepts of physics such as motion, energy, force, time, thermodynamics, quantum chemistry, statistical mechanics, analytical dynamics and chemical equilibria.

In the physical sciences, a phase is a region of material that is chemically uniform, physically distinct, and (often) mechanically separable. In a system consisting of ice and water in a glass jar, the ice cubes are one phase, the water is a second phase, and the humid air is a third phase over the ice and water. The glass of the jar is another separate phase.
Raoult's law ( law) is a relation of physical chemistry, with implications in thermodynamics. Proposed by French chemist François-Marie Raoult in 1887, it states that the partial pressure of each component of an ideal mixture of liquids is equal to the vapor pressure of the pure component multiplied by its mole fraction in the mixture. In consequence, the relative lowering of vapor pressure of a dilute solution of nonvolatile solute is equal to the mole fraction of solute in the solution.

Vapor pressure or equilibrium vapor pressure is the pressure exerted by a vapor in thermodynamic equilibrium with its condensed phases at a given temperature in a closed system. The equilibrium vapor pressure is an indication of a liquid's thermodynamic tendency to evaporate. It relates to the balance of particles escaping from the liquid in equilibrium with those in a coexisting vapor phase. A substance with a high vapor pressure at normal temperatures is often referred to as volatile. The pressure exhibited by vapor present above a liquid surface is known as vapor pressure. As the temperature of a liquid increases, the attractive interactions between liquid molecules become less significant in comparison to the entropy of those molecules in the gas phase, increasing the vapor pressure. Thus, liquids with strong intermolecular interactions are likely to have smaller vapor pressures, with the reverse true for weaker interactions.
In chemical thermodynamics, activity is a measure of the "effective concentration" of a species in a mixture, in the sense that the species' chemical potential depends on the activity of a real solution in the same way that it would depend on concentration for an ideal solution. The term "activity" in this sense was coined by the American chemist Gilbert N. Lewis in 1907.
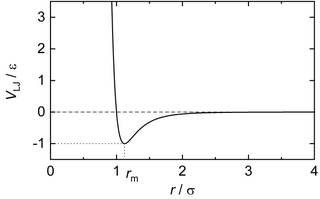
In computational chemistry, molecular physics, and physical chemistry the Lennard-Jones potential is an intermolecular pair potential. Out of all the intermolecular potentials, the Lennard-Jones potential is probably the one that has been the most extensively studied. It is considered an archetype model for simple yet realistic intermolecular interactions.
Chemical kinetics, also known as reaction kinetics, is the branch of physical chemistry that is concerned with understanding the rates of chemical reactions. It is different from chemical thermodynamics, which deals with the direction in which a reaction occurs but in itself tells nothing about its rate. Chemical kinetics includes investigations of how experimental conditions influence the speed of a chemical reaction and yield information about the reaction's mechanism and transition states, as well as the construction of mathematical models that also can describe the characteristics of a chemical reaction.
In chemical thermodynamics, the fugacity of a real gas is an effective partial pressure which replaces the mechanical partial pressure in an accurate computation of chemical equilibrium. It is equal to the pressure of an ideal gas which has the same temperature and molar Gibbs free energy as the real gas.
In thermodynamics, an activity coefficient is a factor used to account for deviation of a mixture of chemical substances from ideal behaviour. In an ideal mixture, the microscopic interactions between each pair of chemical species are the same and, as a result, properties of the mixtures can be expressed directly in terms of simple concentrations or partial pressures of the substances present e.g. Raoult's law. Deviations from ideality are accommodated by modifying the concentration by an activity coefficient. Analogously, expressions involving gases can be adjusted for non-ideality by scaling partial pressures by a fugacity coefficient.
The Dortmund Data Bank is a factual data bank for thermodynamic and thermophysical data. Its main usage is the data supply for process simulation where experimental data are the basis for the design, analysis, synthesis, and optimization of chemical processes. The DDB is used for fitting parameters for thermodynamic models like NRTL or UNIQUAC and for many different equations describing pure component properties, e.g., the Antoine equation for vapor pressures. The DDB is also used for the development and revision of predictive methods like UNIFAC and PSRK.

COSMO is a calculation method for determining the electrostatic interaction of a molecule with a solvent. COSMO is a dielectric continuum model. These models can be used in computational chemistry to model solvation effects. COSMO has become a popular method of these solvation models in recent years. The COSMO formalism is similar to the method proposed earlier by Hoshi et al. The COSMO approach is based - as many other dielectric continuum models - on the surface segmentation of a molecule surface.

In statistical thermodynamics, the UNIFAC method is a semi-empirical system for the prediction of non-electrolyte activity in non-ideal mixtures. UNIFAC uses the functional groups present on the molecules that make up the liquid mixture to calculate activity coefficients. By using interactions for each of the functional groups present on the molecules, as well as some binary interaction coefficients, the activity of each of the solutions can be calculated. This information can be used to obtain information on liquid equilibria, which is useful in many thermodynamic calculations, such as chemical reactor design, and distillation calculations.

The non-random two-liquid model is an activity coefficient model introduced by Renon and Prausnitz in 1968 that correlates the activity coefficients of a compound with its mole fractions in the liquid phase concerned. It is frequently applied in the field of chemical engineering to calculate phase equilibria. The concept of NRTL is based on the hypothesis of Wilson, who stated that the local concentration around a molecule in most mixtures is different from the bulk concentration. This difference is due to a difference between the interaction energy of the central molecule with the molecules of its own kind and that with the molecules of the other kind . The energy difference also introduces a non-randomness at the local molecular level. The NRTL model belongs to the so-called local-composition models. Other models of this type are the Wilson model, the UNIQUAC model, and the group contribution model UNIFAC. These local-composition models are not thermodynamically consistent for a one-fluid model for a real mixture due to the assumption that the local composition around molecule i is independent of the local composition around molecule j. This assumption is not true, as was shown by Flemr in 1976. However, they are consistent if a hypothetical two-liquid model is used. Models, which have consistency between bulk and the local molecular concentrations around different types of molecules are COSMO-RS, and COSMOSPACE.

In statistical thermodynamics, UNIQUAC is an activity coefficient model used in description of phase equilibria. The model is a so-called lattice model and has been derived from a first order approximation of interacting molecule surfaces. The model is, however, not fully thermodynamically consistent due to its two-liquid mixture approach. In this approach the local concentration around one central molecule is assumed to be independent from the local composition around another type of molecule.

A liquid is a nearly incompressible fluid that conforms to the shape of its container but retains a nearly constant volume independent of pressure. It is one of the four fundamental states of matter, and is the only state with a definite volume but no fixed shape.

Diffusion is the net movement of anything generally from a region of higher concentration to a region of lower concentration. Diffusion is driven by a gradient in Gibbs free energy or chemical potential. It is possible to diffuse "uphill" from a region of lower concentration to a region of higher concentration, as in spinodal decomposition. Diffusion is a stochastic process due to the inherent randomness of the diffusing entity and can be used to model many real-life stochastic scenarios. Therefore, diffusion and the corresponding mathematical models are used in several fields beyond physics, such as statistics, probability theory, information theory, neural networks, finance, and marketing.
The Van Laar equation is a thermodynamic activity model, which was developed by Johannes van Laar in 1910-1913, to describe phase equilibria of liquid mixtures. The equation was derived from the Van der Waals equation. The original van der Waals parameters didn't give good description of vapor-liquid equilibria of phases, which forced the user to fit the parameters to experimental results. Because of this, the model lost the connection to molecular properties, and therefore it has to be regarded as an empirical model to correlate experimental results.
COSMO-RS is a quantum chemistry based equilibrium thermodynamics method with the purpose of predicting chemical potentials µ in liquids. It processes the screening charge density σ on the surface of molecules to calculate the chemical potential µ of each species in solution. Perhaps in dilute solution a constant potential must be considered. As an initial step a quantum chemical COSMO calculation for all molecules is performed and the results are stored in a database. In a separate step COSMO-RS uses the stored COSMO results to calculate the chemical potential of the molecules in a liquid solvent or mixture. The resulting chemical potentials are the basis for other thermodynamic equilibrium properties such as activity coefficients, solubility, partition coefficients, vapor pressure and free energy of solvation. The method was developed to provide a general prediction method with no need for system specific adjustment.
John Michael Prausnitz is an emeritus professor of chemical engineering at the University of California, Berkeley.
References
- ↑ Andreas Klamt, Gerard J. P. Krooshof, Ross Taylor "COSMOSPACE: Alternative to conventional activity-coefficient models", AIChE J., 48(10), 2332–2349, (2002)
- ↑ Thesis of Dennis Bosse, "Diffusion, Viscosity, and Thermodynamics in Liquid Systems", Technischen Universität Kaiserslautern (2005), Archived 22 May 2011 at the Wayback Machine "