
In genetics, a single-nucleotide polymorphism is a germline substitution of a single nucleotide at a specific position in the genome. Although certain definitions require the substitution to be present in a sufficiently large fraction of the population, many publications do not apply such a frequency threshold.

The Entrez Global Query Cross-Database Search System is a federated search engine, or web portal that allows users to search many discrete health sciences databases at the National Center for Biotechnology Information (NCBI) website. The NCBI is a part of the National Library of Medicine (NLM), which is itself a department of the National Institutes of Health (NIH), which in turn is a part of the United States Department of Health and Human Services. The name "Entrez" was chosen to reflect the spirit of welcoming the public to search the content available from the NLM.
Scopus is Elsevier's abstract and citation database launched in 2004. Scopus covers nearly 36,377 titles from approximately 11,678 publishers, of which 34,346 are peer-reviewed journals in top-level subject fields: life sciences, social sciences, physical sciences and health sciences. It covers three types of sources: book series, journals, and trade journals. All journals covered in the Scopus database are reviewed for sufficiently high quality each year according to four types of numerical quality measure for each title; those are h-Index, CiteScore, SJR and SNIP. Searches in Scopus also incorporate searches of patent database Lexis-Nexis, albeit with a limited functionaly.
The International HapMap Project was an organization that aimed to develop a haplotype map (HapMap) of the human genome, to describe the common patterns of human genetic variation. HapMap is used to find genetic variants affecting health, disease and responses to drugs and environmental factors. The information produced by the project is made freely available for research.
Comparative genomic hybridization(CGH) is a molecular cytogenetic method for analysing copy number variations (CNVs) relative to ploidy level in the DNA of a test sample compared to a reference sample, without the need for culturing cells. The aim of this technique is to quickly and efficiently compare two genomic DNA samples arising from two sources, which are most often closely related, because it is suspected that they contain differences in terms of either gains or losses of either whole chromosomes or subchromosomal regions. This technique was originally developed for the evaluation of the differences between the chromosomal complements of solid tumor and normal tissue, and has an improved resolution of 5–10 megabases compared to the more traditional cytogenetic analysis techniques of giemsa banding and fluorescence in situ hybridization (FISH) which are limited by the resolution of the microscope utilized.

Loss of heterozygosity (LOH) is a type of genetic abnormality in diploid organisms in which one copy of an entire gene and its surrounding chromosomal region are lost. Since diploid cells have two copies of their genes, one from each parent, a single copy of the lost gene still remains when this happens, but any heterozygosity is no longer present.
In molecular biology, SNP array is a type of DNA microarray which is used to detect polymorphisms within a population. A single nucleotide polymorphism (SNP), a variation at a single site in DNA, is the most frequent type of variation in the genome. Around 335 million SNPs have been identified in the human genome, 15 million of which are present at frequencies of 1% or higher across different populations worldwide.

In genomics, a genome-wide association study, also known as whole genome association study, is an observational study of a genome-wide set of genetic variants in different individuals to see if any variant is associated with a trait. GWA studies typically focus on associations between single-nucleotide polymorphisms (SNPs) and traits like major human diseases, but can equally be applied to any other genetic variants and any other organisms.

The 1000 Genomes Project, launched in January 2008, was an international research effort to establish by far the most detailed catalogue of human genetic variation. Scientists planned to sequence the genomes of at least one thousand anonymous participants from a number of different ethnic groups within the following three years, using newly developed technologies which were faster and less expensive. In 2010, the project finished its pilot phase, which was described in detail in a publication in the journal Nature. In 2012, the sequencing of 1092 genomes was announced in a Nature publication. In 2015, two papers in Nature reported results and the completion of the project and opportunities for future research.
The Illumina Methylation Assay using the Infinium I platform uses 'BeadChip' technology to generate a comprehensive genome-wide profiling of human DNA methylation. Similar to bisulfite sequencing and pyrosequencing, this method quantifies methylation levels at various loci within the genome. This assay is used for methylation probes on the Illumina Infinium HumanMethylation27 BeadChip. Probes on the 27k array target regions of the human genome to measure methylation levels at 27,578 CpG dinucleotides in 14,495 genes. The Infinium HumanMethylation450 BeadChip array targets > 450,000 methylation sites. In 2016, the Infinium MethylationEPIC BeadChip was released, which interrogates over 850,000 methylation sites across the human genome.
Virtual karyotype is the digital information reflecting a karyotype, resulting from the analysis of short sequences of DNA from specific loci all over the genome, which are isolated and enumerated. It detects genomic copy number variations at a higher resolution for level than conventional karyotyping or chromosome-based comparative genomic hybridization (CGH). The main methods used for creating virtual karyotypes are array-comparative genomic hybridization and SNP arrays.

Copy number analysis is the process of analyzing data produced by a test for DNA copy number variation in an organism's sample. One application of such analysis is the detection of chromosomal copy number variation that may cause or may increase risks of various critical disorders. Copy number variation can be detected with various types of tests such as fluorescent in situ hybridization, comparative genomic hybridization and with high-resolution array-based tests based on array comparative genomic hybridization, SNP array technologies and high resolution microarrays that include copy number probes as well an SNPs. Array-based methods have been accepted as the most efficient in terms of their resolution and high-throughput nature and the highest coverage and they are also referred to as virtual karyotype. Data analysis for an array-based DNA copy number test can be very challenging though due to very high volume of data that come out of an array platform.
GeneCards is a database of human genes that provides genomic, proteomic, transcriptomic, genetic and functional information on all known and predicted human genes. It is being developed and maintained by the Crown Human Genome Center at the Weizmann Institute of Science, in collaboration with LifeMap Sciences.
Molecular Inversion Probe (MIP) belongs to the class of Capture by Circularization molecular techniques for performing genomic partitioning, a process through which one captures and enriches specific regions of the genome. Probes used in this technique are single stranded DNA molecules and, similar to other genomic partitioning techniques, contain sequences that are complementary to the target in the genome; these probes hybridize to and capture the genomic target. MIP stands unique from other genomic partitioning strategies in that MIP probes share the common design of two genomic target complementary segments separated by a linker region. With this design, when the probe hybridizes to the target, it undergoes an inversion in configuration and circularizes. Specifically, the two target complementary regions at the 5’ and 3’ ends of the probe become adjacent to one another while the internal linker region forms a free hanging loop. The technology has been used extensively in the HapMap project for large-scale SNP genotyping as well as for studying gene copy alterations and characteristics of specific genomic loci to identify biomarkers for different diseases such as cancer. Key strengths of the MIP technology include its high specificity to the target and its scalability for high-throughput, multiplexed analyses where tens of thousands of genomic loci are assayed simultaneously.
The Functional Element SNPs Database (FESD) is a biological database of single nucleotide polymorphisms in molecular biology. The database is a tool designed to organize functional elements into categories in human gene regions and to output their sequences needed for genotyping experiments as well as provide a set of SNPs that lie within each region. The database defines functional elements into ten types: promoter regions, CpG islands,5' untranslated regions (5'-UTRs), translation start sites, splice sites, coding exons, introns, translation stop sites, polyadenylation signals, and 3' UTRs. People may reference this database for haplotype information or obtain a flanking sequence for genotyping. This may help in finding mutations that contribute to common and polygenic diseases. Researchers can manually choose a group of SNPs of special interest for certain functional elements along with their corresponding sequences. The database combines information from sources such as HapMap, UCSC GoldenPath, dbSNP, OMIM, and TRANSFAC. Users can obtain information about tag SNPs and simulate LD blocks for each gene. FESD is still a developing database and is not widely known so was unable to find projects that used the database. Research was found using similar databases or databases that are combined in FESD’s information pool.
Pan-cancer analysis aims to examine the similarities and differences among the genomic and cellular alterations found across diverse tumor types. International efforts have performed pan-cancer analysis on exomes and the whole genomes of cancers, the latter including their non-coding regions. In 2018, The Cancer Genome Atlas (TCGA) Research Network used exome, transcriptome, and DNA methylome data to develop an integrated picture of commonalities, differences, and emergent themes across tumor types.
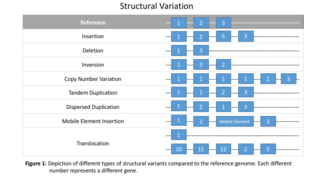
Structural variation in the human genome is operationally defined as genomic alterations, varying between individuals, that involve DNA segments larger than 1 kilo base (kb), and could be either microscopic or submicroscopic. This definition distinguishes them from smaller variants that are less than 1 kb in size such as short deletions, insertions, and single nucleotide variants.
The GWAS catalog is a free online database that compiles data of genome-wide association studies (GWAS), summarizing unstructured data from different literature sources into accessible high quality data. It was created by the National Human Genome Research Institute (NHGRI) in 2008 and have become a collaborative project between the NHGRI and the European Bioinformatics Institute (EBI) since 2010. As of September 2018, it has included 71,673 SNP–trait associations in 3,567 publications.