
Proteasomes are protein complexes which degrade unneeded or damaged proteins by proteolysis, a chemical reaction that breaks peptide bonds. Enzymes that help such reactions are called proteases.
In molecular biology and pharmacology, a small molecule or micromolecule is a low molecular weight organic compound that may regulate a biological process, with a size on the order of 1 nm. Many drugs are small molecules; the terms are equivalent in the literature. Larger structures such as nucleic acids and proteins, and many polysaccharides are not small molecules, although their constituent monomers are often considered small molecules. Small molecules may be used as research tools to probe biological function as well as leads in the development of new therapeutic agents. Some can inhibit a specific function of a protein or disrupt protein–protein interactions.

In biochemistry and molecular biology, a binding site is a region on a macromolecule such as a protein that binds to another molecule with specificity. The binding partner of the macromolecule is often referred to as a ligand. Ligands may include other proteins, enzyme substrates, second messengers, hormones, or allosteric modulators. The binding event is often, but not always, accompanied by a conformational change that alters the protein's function. Binding to protein binding sites is most often reversible, but can also be covalent reversible or irreversible.
A hormone receptor is a receptor molecule that binds to a specific hormone. Hormone receptors are a wide family of proteins made up of receptors for thyroid and steroid hormones, retinoids and Vitamin D, and a variety of other receptors for various ligands, such as fatty acids and prostaglandins. Hormone receptors are of mainly two classes. Receptors for peptide hormones tend to be cell surface receptors built into the plasma membrane of cells and are thus referred to as trans membrane receptors. An example of this is Actrapid. Receptors for steroid hormones are usually found within the protoplasm and are referred to as intracellular or nuclear receptors, such as testosterone. Upon hormone binding, the receptor can initiate multiple signaling pathways, which ultimately leads to changes in the behavior of the target cells.

A ubiquitin ligase is a protein that recruits an E2 ubiquitin-conjugating enzyme that has been loaded with ubiquitin, recognizes a protein substrate, and assists or directly catalyzes the transfer of ubiquitin from the E2 to the protein substrate. In simple and more general terms, the ligase enables movement of ubiquitin from a ubiquitin carrier to another thing by some mechanism. The ubiquitin, once it reaches its destination, ends up being attached by an isopeptide bond to a lysine residue, which is part of the target protein. E3 ligases interact with both the target protein and the E2 enzyme, and so impart substrate specificity to the E2. Commonly, E3s polyubiquitinate their substrate with Lys48-linked chains of ubiquitin, targeting the substrate for destruction by the proteasome. However, many other types of linkages are possible and alter a protein's activity, interactions, or localization. Ubiquitination by E3 ligases regulates diverse areas such as cell trafficking, DNA repair, and signaling and is of profound importance in cell biology. E3 ligases are also key players in cell cycle control, mediating the degradation of cyclins, as well as cyclin dependent kinase inhibitor proteins. The human genome encodes over 600 putative E3 ligases, allowing for tremendous diversity in substrates.

P-glycoprotein 1 also known as multidrug resistance protein 1 (MDR1) or ATP-binding cassette sub-family B member 1 (ABCB1) or cluster of differentiation 243 (CD243) is an important protein of the cell membrane that pumps many foreign substances out of cells. More formally, it is an ATP-dependent efflux pump with broad substrate specificity. It exists in animals, fungi, and bacteria, and it likely evolved as a defense mechanism against harmful substances.
Heterogeneous nuclear ribonucleoproteins (hnRNPs) are complexes of RNA and protein present in the cell nucleus during gene transcription and subsequent post-transcriptional modification of the newly synthesized RNA (pre-mRNA). The presence of the proteins bound to a pre-mRNA molecule serves as a signal that the pre-mRNA is not yet fully processed and therefore not ready for export to the cytoplasm. Since most mature RNA is exported from the nucleus relatively quickly, most RNA-binding protein in the nucleus exist as heterogeneous ribonucleoprotein particles. After splicing has occurred, the proteins remain bound to spliced introns and target them for degradation.
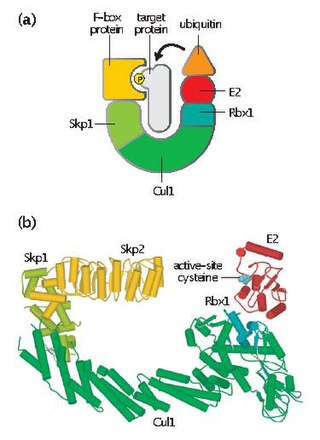
Skp, Cullin, F-box containing complex is a multi-protein E3 ubiquitin ligase complex that catalyzes the ubiquitination of proteins destined for 26S proteasomal degradation. Along with the anaphase-promoting complex, SCF has important roles in the ubiquitination of proteins involved in the cell cycle. The SCF complex also marks various other cellular proteins for destruction.
Phosphodiesterase 1, PDE1, EC 3.1.4.1, systematic name oligonucleotide 5′-nucleotidohydrolase) is a phosphodiesterase enzyme also known as calcium- and calmodulin-dependent phosphodiesterase. It is one of the 11 families of phosphodiesterase (PDE1-PDE11). Phosphodiesterase 1 has three subtypes, PDE1A, PDE1B and PDE1C which divide further into various isoforms. The various isoforms exhibit different affinities for cAMP and cGMP.

Fusion proteins or chimeric (kī-ˈmir-ik) proteins are proteins created through the joining of two or more genes that originally coded for separate proteins. Translation of this fusion gene results in a single or multiple polypeptides with functional properties derived from each of the original proteins. Recombinant fusion proteins are created artificially by recombinant DNA technology for use in biological research or therapeutics. Chimeric or chimera usually designate hybrid proteins made of polypeptides having different functions or physico-chemical patterns. Chimeric mutant proteins occur naturally when a complex mutation, such as a chromosomal translocation, tandem duplication, or retrotransposition creates a novel coding sequence containing parts of the coding sequences from two different genes. Naturally occurring fusion proteins are commonly found in cancer cells, where they may function as oncoproteins. The bcr-abl fusion protein is a well-known example of an oncogenic fusion protein, and is considered to be the primary oncogenic driver of chronic myelogenous leukemia.
The unfolded protein response (UPR) is a cellular stress response related to the endoplasmic reticulum (ER) stress. It has been found to be conserved between mammalian species, as well as yeast and worm organisms.
Proteostasis is the dynamic regulation of a balanced, functional proteome. The proteostasis network includes competing and integrated biological pathways within cells that control the biogenesis, folding, trafficking, and degradation of proteins present within and outside the cell. Loss of proteostasis is central to understanding the cause of diseases associated with excessive protein misfolding and degradation leading to loss-of-function phenotypes, as well as aggregation-associated degenerative disorders. Therapeutic restoration of proteostasis may treat or resolve these pathologies.

An Hsp90 inhibitor is a substance that inhibits that activity of the Hsp90 heat shock protein. Since Hsp90 stabilizes a variety of proteins required for survival of cancer cells, these substances may have therapeutic benefit in the treatment of various types of malignancies. Furthermore, a number of Hsp90 inhibitors are currently undergoing clinical trials for a variety of cancers. Hsp90 inhibitors include the natural products geldanamycin and radicicol as well as semisynthetic derivatives 17-N-Allylamino-17-demethoxygeldanamycin (17AAG).
A selective estrogen receptor degrader or downregulator (SERD) is a type of drug which binds to the estrogen receptor (ER) and, in the process of doing so, causes the ER to be degraded and thus downregulated. They are used to treat estrogen receptor-sensitive or progesterone receptor-sensitive breast cancer, along with older classes of drugs like selective estrogen receptor modulators (SERMs) and aromatase inhibitors.
Kang-Yell Choi is a professor of biotechnology at Yonsei University, and has a joint appointment position as a CEO of CK Regeon Inc. in Seoul, Korea. He has been performing researches related to cellular signaling, especially for the Wnt/β-catenin pathway involving various pathophysiologies. Choi has been leading the Translational Research Center for Protein Function Control (TRCP), a Korean government supported drug development institute, as a director for 10 years. Choi has been carrying out R&D to develop agents controlling the Wnt/β-catenin signaling pathway. Choi's main interest is development of the agents to treat intractable diseases that suppress tissue regeneration system through overexpression of CXXC5 and subsequent suppression of the Wnt/β-catenin signaling.
Craig M. Crews is an American scientist at Yale University known for his contributions to chemical biology. He is known for his contributions to the field of induced proximity through his work in creating heterobifunctional molecules that hijack cellular processes by inducing the interaction of two proteins inside a living cell. His initial work focused on the discovery of PROteolysis-TArgeting Chimeras (PROTACs) to trigger degradation of disease-causing proteins, a process known as targeted protein degradation (TPD), and he has since developed new versions of -TACs to leverage other cellular processes and protein families to treat disease.
A proteolysis targeting chimera (PROTAC) is a heterobifunctional molecule composed of two active domains and a linker, capable of removing specific unwanted proteins. Rather than acting as a conventional enzyme inhibitor, a PROTAC works by inducing selective intracellular proteolysis. PROTACs consist of two covalently linked protein-binding molecules: one capable of engaging an E3 ubiquitin ligase, and another that binds to a target protein meant for degradation. Recruitment of the E3 ligase to the target protein results in ubiquitination and subsequent degradation of the target protein via the proteasome. Because PROTACs need only to bind their targets with high selectivity, there are currently many efforts to retool previously ineffective inhibitor molecules as PROTACs for next-generation drugs.

Daniel K. Nomura is an American chemical biologist and Professor of Chemical Biology and Molecular Therapeutics at the University of California, Berkeley, in the Departments of Chemistry and Molecular & Cell Biology. His work employs chemoproteomic approaches to develop small molecule therapeutics and therapeutic modalities against traditionally "undruggable" proteins.
A molecular glue is a small molecule that induces the interaction between two proteins that do not normally interact.
James Allen Wells is a Professor of Pharmaceutical Chemistry and Cellular & Molecular Pharmacology at the University of California, San Francisco (UCSF) and a member of the National Academy of Sciences. He received his B.A. degrees in biochemistry and psychology from University of California, Berkeley in 1973 and a PhD in biochemistry from Washington State University with Ralph Yount, PhD in 1979. He completed his postdoctoral studies at Stanford University School of Medicine with George Stark in 1982. He is a pioneer in protein engineering, phage display, fragment-based lead discovery, cellular apoptosis, and the cell surface proteome.
