Related Research Articles
Clinical trials are experiments or observations done in clinical research. Such prospective biomedical or behavioral research studies on human participants are designed to answer specific questions about biomedical or behavioral interventions, including new treatments and known interventions that warrant further study and comparison. Clinical trials generate data on dosage, safety and efficacy. They are conducted only after they have received health authority/ethics committee approval in the country where approval of the therapy is sought. These authorities are responsible for vetting the risk/benefit ratio of the trial—their approval does not mean the therapy is 'safe' or effective, only that the trial may be conducted.

Bioequivalence is a term in pharmacokinetics used to assess the expected in vivo biological equivalence of two proprietary preparations of a drug. If two products are said to be bioequivalent it means that they would be expected to be, for all intents and purposes, the same.

The Common Technical Document (CTD) is a set of specifications for an application dossier for the registration of Medicines and designed to be used across Europe, Japan and the United States and beyond.
The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) is an initiative that brings together regulatory authorities and pharmaceutical industry to discuss scientific and technical aspects of pharmaceutical product development and registration.
An institutional review board (IRB), also known as an independent ethics committee (IEC), ethical review board (ERB), or research ethics board (REB), is a type of committee that applies research ethics by reviewing the methods proposed for research to ensure that they are ethical. Such boards are formally designated to approve, monitor, and review biomedical and behavioral research involving humans. They often conduct some form of risk-benefit analysis in an attempt to determine whether or not research should be conducted. The purpose of the IRB is to assure that appropriate steps are taken to protect the rights and welfare of humans participating as subjects in a research study. Along with developed countries, many developing countries have established national, regional or local Institutional Review Boards in order to safeguard ethical conduct of research concerning both national and international norms, regulations or codes.
The standard treatment, also known as the standard of care, is the medical treatment that is normally provided to people with a given condition. In many scientific studies, the control group receives the standard treatment rather than a placebo while a treatment group receives the experimental treatment. After the clinical trial, researchers compare the outcomes of the two groups to see if the experimental treatment is better than, as good as or not as beneficial as the standard treatment.
A serious adverse event (SAE) in human drug trials is defined as any untoward medical occurrence that at any dose
- Results in death
- Is life-threatening
- Requires inpatient hospitalization or causes prolongation of existing hospitalization
- Results in persistent or significant disability/incapacity
- May have caused a congenital anomaly/birth defect
- Requires intervention to prevent permanent impairment or damage
Therapeutic Products Directorate (TPD) is a Canadian federal authority that regulates pharmaceutical drugs for human use. Prior to being given market authorization, a manufacturer must present substantive scientific evidence of a product's safety, efficacy, and quality as required by the Food and Drugs Act and Regulations. It is one of the seven operational directorates of the Health Products and Food Branch, a branch of Health Canada.
Good clinical practice (GCP) is an international quality standard, which governments can then transpose into regulations for clinical trials involving human subjects. GCP follows the International Council on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), and enforces tight guidelines on ethical aspects of clinical research.
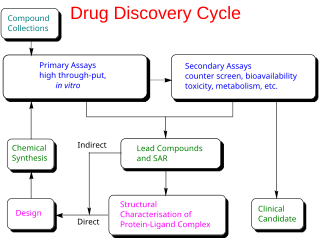
Drug development is the process of bringing a new pharmaceutical drug to the market once a lead compound has been identified through the process of drug discovery. It includes preclinical research on microorganisms and animals, filing for regulatory status, such as via the United States Food and Drug Administration for an investigational new drug to initiate clinical trials on humans, and may include the step of obtaining regulatory approval with a new drug application to market the drug. The entire process – from concept through preclinical testing in the laboratory to clinical trial development, including Phase I–III trials – to approved vaccine or drug typically takes more than a decade.
In drug development and medical device development the Investigator's Brochure (IB) is a comprehensive document summarizing the body of information about an investigational product obtained during a drug trial. The IB is a document of critical importance throughout the drug development process and is updated with new information as it becomes available. The purpose of the IB is to compile data relevant to studies of the IP in human subjects gathered during preclinical and other clinical trials.
A clinical investigator involved in a clinical trial is responsible for ensuring that an investigation is conducted according to the signed investigator statement, the investigational plan, and applicable regulations; for protecting the rights, safety, and welfare of subjects under the investigator's care; and for the control of drugs under investigation. The Clinical Investigator must also meet requirements set forth by the FDA, EMA or other regulatory body. The qualifications must be outlined in a current resume and readily available for auditors.
A Clinical Research Coordinator (CRC) is a person responsible for conducting clinical trials using good clinical practice (GCP) under the auspices of a Principal Investigator (PI).
The analysis of clinical trials involves many related topics including:
A glossary of terms used in clinical research.
The following outline is provided as an overview of and topical guide to clinical research:
An estimand is a quantity that is to be estimated in a statistical analysis. The term is used to more clearly distinguish the target of inference from the method used to obtain an approximation of this target and the specific value obtained from a given method and dataset. For instance, a normally distributed random variable has two defining parameters, its mean and variance . A variance estimator:
In medicine, a clinical study report (CSR) on a clinical trial is a document, typically very long, providing much detail about the methods and results of a trial. A CSR is a scientific document addressing efficacy and safety, not a sales or marketing tool; its content is similar to that of a peer-reviewed academic paper. Results of trials are usually reported in a briefer academic journal paper, but methodological flaws are often glossed over in the briefer paper.
Guidances for statistics in regulatory affairs are applicable to the pharmaceutical industry and medical devices industry. These Guidances represent the current thinking of regulatory agencies on a particular subject. It is to be noted that the term “Guidances” is used in the USA, whereas the term “Guidelines” is used in Europe.
References
- ↑ Statistical Principles for Clinical Trials (PDF). International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH. 1998-02-05. Archived from the original (PDF) on 2008-09-21.