Related Research Articles

Hemoglobinopathy is the medical term for a group of inherited blood disorders and diseases that primarily affect red blood cells. They are single-gene disorders and, in most cases, they are inherited as autosomal co-dominant traits.

Anemia or anaemia is a blood disorder in which the blood has a reduced ability to carry oxygen due to a lower than normal number of red blood cells, or a reduction in the amount of hemoglobin. When anemia comes on slowly, the symptoms are often vague, such as tiredness, weakness, shortness of breath, headaches, and a reduced ability to exercise. When anemia is acute, symptoms may include confusion, feeling like one is going to pass out, loss of consciousness, and increased thirst. Anemia must be significant before a person becomes noticeably pale. Additional symptoms may occur depending on the underlying cause. Preoperative anemia can increase the risk of needing a blood transfusion following surgery. Anemia can be temporary or long term and can range from mild to severe.

A myelodysplastic syndrome (MDS) is one of a group of cancers in which immature blood cells in the bone marrow do not mature, so do not become healthy blood cells. Early on, no symptoms typically are seen. Later, symptoms may include feeling tired, shortness of breath, bleeding disorders, anemia, or frequent infections. Some types may develop into acute myeloid leukemia.

Thalassemias are inherited blood disorders characterized by decreased hemoglobin production. Symptoms depend on the type and can vary from none to severe. Often there is mild to severe anemia. Anemia can result in feeling tired and pale skin. There may also be bone problems, an enlarged spleen, yellowish skin, and dark urine. Slow growth may occur in children.

Chelation therapy is a medical procedure that involves the administration of chelating agents to remove heavy metals from the body. Chelation therapy has a long history of use in clinical toxicology and remains in use for some very specific medical treatments, although it is administered under very careful medical supervision due to various inherent risks, including the mobilization of mercury and other metals through the brain and other parts of the body by the use of weak chelating agents that unbind with metals before elimination, exacerbating existing damage. To avoid mobilization, some practitioners of chelation use strong chelators, such as selenium, taken at low doses over a long period of time.
Deferoxamine (DFOA), also known as desferrioxamine and sold under the brand name Desferal, is a medication that binds iron and aluminium. It is specifically used in iron overdose, hemochromatosis either due to multiple blood transfusions or an underlying genetic condition, and aluminium toxicity in people on dialysis. It is used by injection into a muscle, vein, or under the skin.

Hepcidin is a protein that in humans is encoded by the HAMP gene. Hepcidin is a key regulator of the entry of iron into the circulation in mammals.

Alpha-thalassemia is a form of thalassemia involving the genes HBA1 and HBA2. Thalassemias are a group of inherited blood conditions which result in the impaired production of hemoglobin, the molecule that carries oxygen in the blood. Normal hemoglobin consists of two alpha chains and two beta chains; in alpha-thalassemia, there is a quantitative decrease in the amount of alpha chains, resulting in fewer normal hemoglobin molecules. Furthermore, alpha-thalassemia leads to the production of unstable beta globin molecules which cause increased red blood cell destruction. The degree of impairment is based on which clinical phenotype is present.

Hypochromic anemia is a generic term for any type of anemia in which the red blood cells are paler than normal. A normal red blood cell has a biconcave disk shape and will have an area of pallor in its center when viewed microscopically. In hypochromic cells, this area of central pallor is increased. This decrease in redness is due to a disproportionate reduction of red cell hemoglobin in proportion to the volume of the cell. Clinically the color can be evaluated by the mean corpuscular hemoglobin (MCH) or mean corpuscular hemoglobin concentration (MCHC). The MCHC is considered the better parameter of the two as it adjusts for effect the size of the cell has on its amount of hemoglobin. Hypochromia is clinically defined as below the normal MCH reference range of 27–33 picograms/cell in adults or below the normal MCHC reference range of 33–36 g/dL in adults.
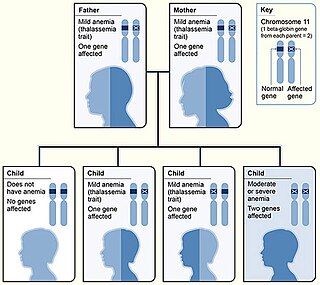
Beta thalassemias are a group of inherited blood disorders. They are forms of thalassemia caused by reduced or absent synthesis of the beta chains of hemoglobin that result in variable outcomes ranging from severe anemia to clinically asymptomatic individuals. Global annual incidence is estimated at one in 100,000. Beta thalassemias occur due to malfunctions in the hemoglobin subunit beta or HBB. The severity of the disease depends on the nature of the mutation.
Nancy Fern Olivieri is a prominent Toronto haematologist and researcher with an interest in the treatment of haemoglobinopathies. She is best known for a protracted struggle with the Hospital for Sick Children and the pharmaceutical company Apotex about the drug deferiprone.

Deferiprone, sold under the brand name Ferriprox among others, is a medication that chelates iron and is used to treat iron overload in thalassaemia major. It was first approved and indicated for use in treating thalassaemia major in 1994 and had been licensed for use in the European Union and in Asia for many years while awaiting approval in Canada and in the United States. On October 14, 2011, it was approved for use in the US under the FDA's accelerated approval program.

Hemosiderosis is a form of iron overload disorder resulting in the accumulation of hemosiderin.

Congenital dyserythropoietic anemia (CDA) is a rare blood disorder, similar to the thalassemias. CDA is one of many types of anemia, characterized by ineffective erythropoiesis, and resulting from a decrease in the number of red blood cells (RBCs) in the body and a less than normal quantity of hemoglobin in the blood. CDA may be transmitted by both parents autosomal recessively or dominantly.
Treatment of the inherited blood disorder thalassemia depends upon the level of severity. For mild forms of the condition, advice and counseling are often all that are necessary. For more severe forms, treatment may consist in blood transfusion; chelation therapy to reverse iron overload, using drugs such as deferoxamine, deferiprone, or deferasirox; medication with the antioxidant indicaxanthin to prevent the breakdown of hemoglobin; or a bone marrow transplant using material from a compatible donor, or from the patient's mother. Removal of the spleen (splenectomy) could theoretically help to reduce the need for blood transfusions in people with thalassaemia major or intermedia but there is currently no reliable evidence from clinical trials about its effects. Population screening has had some success as a preventive measure.
Voluntary Health Services, popularly known as the VHS Hospital, is a multispecialty tertiary care referral hospital in the south Indian state of Tamil Nadu, reportedly serving the economically weaker sections of the society. It was founded in 1958 by Krishnaswami Srinivas Sanjivi, an Indian physician, social worker and a winner of Padma Shri and Padma Bhushan awards and is run by a charitable non governmental organization of the same name. The hospital is situated along Rajiv Gandhi Salai at Taramani, in Chennai.
Jan Olav Aaseth is a Norwegian physician, doctor of philosophy, professor, research advisor in internal medicine, endocrinology, toxicology, and medical biochemistry, and advisor for international scientific journals.

Marina Cavazzana is a professor of Paediatric Immunology at the Necker-Enfants Malades Hospital and the Imagine Institute, as well as an academic at Paris Descartes University. She was awarded the Irène Joliot-Curie Prize in 2012 and elected to the National Academy of Medicine in 2019.

Transfusion-dependent anemia is a form of anemia characterized by the need for continuous blood transfusion. It is a condition that results from various diseases, and is associated with decreased survival rates. Regular transfusion is required to reduce the symptoms of anemia by increasing functional red blood cells and hemoglobin count. Symptoms may vary based on the severity of the condition and the most common symptom is fatigue. Various diseases can lead to transfusion-dependent anemia, most notably myelodysplastic syndromes (MDS) and thalassemia. Due to the number of diseases that can cause transfusion-dependent anemia, diagnosing it is more complicated. Transfusion dependence occurs when an average of more than 2 units of blood transfused every 28 days is required over a period of at least 3 months. Myelodysplastic syndromes is often only diagnosed when patients become anemic, and transfusion-dependent thalassemia is diagnosed based on gene mutations. Screening for heterozygosity in the thalassemia gene is an option for early detection.
References
- ↑ Emrich, Robert (16 April 2009). "Working Toward a Cure". Queens Chronicle. Retrieved 2 December 2014.
- ↑ "Cooleys Anemia Foundation, Inc. - Research Program (Thalassemia)". researchfunding.duke.edu. Archived from the original on 23 December 2014. Retrieved 2 December 2014.
- ↑ "CAF Medical research Funding 2014" . Retrieved 23 December 2014.
- ↑ Suto, R (15 September 1995). "A mechanism for the specific immunogenicity of heat shock protein-chaperoned peptides". Science. 259 (5230): 1585–1588. doi:10.1126/science.7545313. PMID 7545313.
- ↑ Bergeron, Raymond J.; Brittenham, Gary M. (6 December 1993). The Development of Iron Chelators for Clinical Use. CRC Press. ISBN 978-0849386794 . Retrieved 2 December 2014.
- ↑ "Annals of the New York Academy of Sciences Online Library". Annals of the New York Academy of Sciences. 119 (2). 1964. doi:10.1111/nyas.1964.119.issue-2.
- ↑ "ClinicalTrials.gov TCRN listing" . Retrieved 23 December 2014.
- ↑ "CDC Thalassemia Data Collection and Blood Safety Monitoring page" . Retrieved 23 December 2014.