Related Research Articles

Bioinformatics is an interdisciplinary field of science that develops methods and software tools for understanding biological data, especially when the data sets are large and complex. Bioinformatics uses biology, chemistry, physics, computer science, computer programming, information engineering, mathematics and statistics to analyze and interpret biological data. The subsequent process of analyzing and interpreting data is referred to as computational biology.

A protein family is a group of evolutionarily related proteins. In many cases, a protein family has a corresponding gene family, in which each gene encodes a corresponding protein with a 1:1 relationship. The term "protein family" should not be confused with family as it is used in taxonomy.
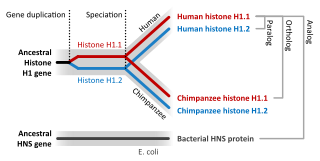
Sequence homology is the biological homology between DNA, RNA, or protein sequences, defined in terms of shared ancestry in the evolutionary history of life. Two segments of DNA can have shared ancestry because of three phenomena: either a speciation event (orthologs), or a duplication event (paralogs), or else a horizontal gene transfer event (xenologs).
The Gene Ontology (GO) is a major bioinformatics initiative to unify the representation of gene and gene product attributes across all species. More specifically, the project aims to: 1) maintain and develop its controlled vocabulary of gene and gene product attributes; 2) annotate genes and gene products, and assimilate and disseminate annotation data; and 3) provide tools for easy access to all aspects of the data provided by the project, and to enable functional interpretation of experimental data using the GO, for example via enrichment analysis. GO is part of a larger classification effort, the Open Biomedical Ontologies, being one of the Initial Candidate Members of the OBO Foundry.

UniProt is a freely accessible database of protein sequence and functional information, many entries being derived from genome sequencing projects. It contains a large amount of information about the biological function of proteins derived from the research literature. It is maintained by the UniProt consortium, which consists of several European bioinformatics organisations and a foundation from Washington, DC, United States.

A fusion gene is a hybrid gene formed from two previously independent genes. It can occur as a result of translocation, interstitial deletion, or chromosomal inversion. Fusion genes have been found to be prevalent in all main types of human neoplasia. The identification of these fusion genes play a prominent role in being a diagnostic and prognostic marker.
The Rat Genome Database (RGD) is a database of rat genomics, genetics, physiology and functional data, as well as data for comparative genomics between rat, human and mouse. RGD is responsible for attaching biological information to the rat genome via structured vocabulary, or ontology, annotations assigned to genes and quantitative trait loci (QTL), and for consolidating rat strain data and making it available to the research community. They are also developing a suite of tools for mining and analyzing genomic, physiologic and functional data for the rat, and comparative data for rat, mouse, human, and five other species.

MicrobesOnline is a publicly and freely accessible website that hosts multiple comparative genomic tools for comparing microbial species at the genomic, transcriptomic and functional levels. MicrobesOnline was developed by the Virtual Institute for Microbial Stress and Survival, which is based at the Lawrence Berkeley National Laboratory in Berkeley, California. The site was launched in 2005, with regular updates until 2011.
SUPERFAMILY is a database and search platform of structural and functional annotation for all proteins and genomes. It classifies amino acid sequences into known structural domains, especially into SCOP superfamilies. Domains are functional, structural, and evolutionary units that form proteins. Domains of common Ancestry are grouped into superfamilies. The domains and domain superfamilies are defined and described in SCOP. Superfamilies are groups of proteins which have structural evidence to support a common evolutionary ancestor but may not have detectable sequence homology.

In molecular biology and genetics, DNA annotation or genome annotation is the process of describing the structure and function of the components of a genome, by analyzing and interpreting them in order to extract their biological significance and understand the biological processes in which they participate. Among other things, it identifies the locations of genes and all the coding regions in a genome and determines what those genes do.
Blast2GO, first published in 2005, is a bioinformatics software tool for the automatic, high-throughput functional annotation of novel sequence data. It makes use of the BLAST algorithm to identify similar sequences to then transfers existing functional annotation from yet characterised sequences to the novel one. The functional information is represented via the Gene Ontology (GO), a controlled vocabulary of functional attributes. The Gene Ontology, or GO, is a major bioinformatics initiative to unify the representation of gene and gene product attributes across all species.
BASys is a freely available web server that can be used to perform automated, comprehensive annotation of bacterial genomes. With the advent of next generation DNA sequencing it is now possible to sequence the complete genome of a bacterium within a single day. This has led to an explosion in the number of fully sequenced microbes. In fact, as of 2013, there were more than 2700 fully sequenced bacterial genomes deposited with GenBank. However, a continuing challenge with microbial genomics is finding the resources or tools for annotating the large number of newly sequenced genomes. BASys was developed in 2005 in anticipation of these needs. In fact, BASys was the world’s first publicly accessible microbial genome annotation web server. Because of its widespread popularity, the BASys server was updated in 2011 through the addition of multiple server nodes to handle the large number of queries it was receiving.

Gene set enrichment analysis (GSEA) (also called functional enrichment analysis or pathway enrichment analysis) is a method to identify classes of genes or proteins that are over-represented in a large set of genes or proteins, and may have an association with different phenotypes (e.g. different organism growth patterns or diseases). The method uses statistical approaches to identify significantly enriched or depleted groups of genes. Transcriptomics technologies and proteomics results often identify thousands of genes, which are used for the analysis.
In bioinformatics, a Gene Disease Database is a systematized collection of data, typically structured to model aspects of reality, in a way to comprehend the underlying mechanisms of complex diseases, by understanding multiple composite interactions between phenotype-genotype relationships and gene-disease mechanisms. Gene Disease Databases integrate human gene-disease associations from various expert curated databases and text mining derived associations including Mendelian, complex and environmental diseases.
Single nucleotide polymorphism annotation is the process of predicting the effect or function of an individual SNP using SNP annotation tools. In SNP annotation the biological information is extracted, collected and displayed in a clear form amenable to query. SNP functional annotation is typically performed based on the available information on nucleic acid and protein sequences.

Pathway is the term from molecular biology for a curated schematic representation of a well characterized segment of the molecular physiological machinery, such as a metabolic pathway describing an enzymatic process within a cell or tissue or a signaling pathway model representing a regulatory process that might, in its turn, enable a metabolic or another regulatory process downstream. A typical pathway model starts with an extracellular signaling molecule that activates a specific receptor, thus triggering a chain of molecular interactions. A pathway is most often represented as a relatively small graph with gene, protein, and/or small molecule nodes connected by edges of known functional relations. While a simpler pathway might appear as a chain, complex pathway topologies with loops and alternative routes are much more common. Computational analyses employ special formats of pathway representation. In the simplest form, however, a pathway might be represented as a list of member molecules with order and relations unspecified. Such a representation, generally called Functional Gene Set (FGS), can also refer to other functionally characterised groups such as protein families, Gene Ontology (GO) and Disease Ontology (DO) terms etc. In bioinformatics, methods of pathway analysis might be used to identify key genes/ proteins within a previously known pathway in relation to a particular experiment / pathological condition or building a pathway de novo from proteins that have been identified as key affected elements. By examining changes in e.g. gene expression in a pathway, its biological activity can be explored. However most frequently, pathway analysis refers to a method of initial characterization and interpretation of an experimental condition that was studied with omics tools or genome-wide association study. Such studies might identify long lists of altered genes. A visual inspection is then challenging and the information is hard to summarize, since the altered genes map to a broad range of pathways, processes, and molecular functions. In such situations, the most productive way of exploring the list is to identify enrichment of specific FGSs in it. The general approach of enrichment analyses is to identify FGSs, members of which were most frequently or most strongly altered in the given condition, in comparison to a gene set sampled by chance. In other words, enrichment can map canonical prior knowledge structured in the form of FGSs to the condition represented by altered genes.
Metascape is a free gene annotation and analysis resource that helps biologists make sense of one or multiple gene lists. Metascape provides automated meta-analysis tools to understand either common or unique pathways and protein networks within a group of orthogonal target-discovery studies.
Machine learning in bioinformatics is the application of machine learning algorithms to bioinformatics, including genomics, proteomics, microarrays, systems biology, evolution, and text mining.
Biocuration is the field of life sciences dedicated to organizing biomedical data, information and knowledge into structured formats, such as spreadsheets, tables and knowledge graphs. The biocuration of biomedical knowledge is made possible by the cooperative work of biocurators, software developers and bioinformaticians and is at the base of the work of biological databases.
References
- ↑ "License: Users from academic, non-profit and government institutes may freely use EASE". Archived from the original on 2011-10-15. Retrieved 2012-02-04.
- ↑ Huang DW, Lempicki RA (2009). "Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources". Nature Protocols. 4 (1): 44–57. doi:10.1038/nprot.2008.211. PMID 19131956. S2CID 10418677.
- ↑ Sherman BT, Huang DW, Tan Q, Guo Y, Bour S, Liu D, Stephens R, Baseler MW, Lane HC, Lempicki RA (2007). "DAVID Knowledgebase: a gene-centered database integrating heterogeneous gene annotation resources to facilitate high-throughput gene functional analysis". BMC Bioinformatics. 8: 426. doi: 10.1186/1471-2105-8-426 . PMC 2186358 . PMID 17980028.
- ↑ Huang DW, Sherman BT, Tan Q, Collins JR, Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC, Lempicki RA (2007). "The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists". Genome Biol. 8 (9): R183. doi: 10.1186/gb-2007-8-9-r183 . PMC 2375021 . PMID 17784955.
- ↑ Huang Da, HC; Sherman, BT; Tan, Q; Kir, J; Liu, D; Bryant, D; Guo, Y; Stephens, R; et al. (2007). "DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists". Nucleic Acids Research. 35 (Web Server issue): W169–75. doi:10.1093/nar/gkm415. PMC 1933169 . PMID 17576678.
- ↑ Hosack; Dennis Jr, G; Sherman, BT; Lane, HC; Lempicki, RA (2003). "Identifying biological themes within lists of genes with EASE". Genome Biology. 4 (10): R70. doi: 10.1186/gb-2003-4-10-r70 . PMC 328459 . PMID 14519205.
- ↑ Dennis Jr; Sherman, BT; Hosack, DA; Yang, J; Gao, W; Lane, HC; Lempicki, RA (2003). "DAVID: Database for Annotation, Visualization, and Integrated Discovery". Genome Biology. 4 (5): P3. doi: 10.1186/gb-2003-4-5-p3 . PMC 193660 . PMID 12734009.
- ↑ Release & Version Information, DAVID Bioinformatics Resources.