The genotype of an organism is its complete set of genetic material. Genotype can also be used to refer to the alleles or variants an individual carries in a particular gene or genetic location. The number of alleles an individual can have in a specific gene depends on the number of copies of each chromosome found in that species, also referred to as ploidy. In diploid species like humans, two full sets of chromosomes are present, meaning each individual has two alleles for any given gene. If both alleles are the same, the genotype is referred to as homozygous. If the alleles are different, the genotype is referred to as heterozygous.
In molecular biology, restriction fragment length polymorphism (RFLP) is a technique that exploits variations in homologous DNA sequences, known as polymorphisms, populations, or species or to pinpoint the locations of genes within a sequence. The term may refer to a polymorphism itself, as detected through the differing locations of restriction enzyme sites, or to a related laboratory technique by which such differences can be illustrated. In RFLP analysis, a DNA sample is digested into fragments by one or more restriction enzymes, and the resulting restriction fragments are then separated by gel electrophoresis according to their size.

The human genome is a complete set of nucleic acid sequences for humans, encoded as DNA within the 23 chromosome pairs in cell nuclei and in a small DNA molecule found within individual mitochondria. These are usually treated separately as the nuclear genome and the mitochondrial genome. Human genomes include both protein-coding DNA sequences and various types of DNA that does not encode proteins. The latter is a diverse category that includes DNA coding for non-translated RNA, such as that for ribosomal RNA, transfer RNA, ribozymes, small nuclear RNAs, and several types of regulatory RNAs. It also includes promoters and their associated gene-regulatory elements, DNA playing structural and replicatory roles, such as scaffolding regions, telomeres, centromeres, and origins of replication, plus large numbers of transposable elements, inserted viral DNA, non-functional pseudogenes and simple, highly repetitive sequences. Introns make up a large percentage of non-coding DNA. Some of this non-coding DNA is non-functional junk DNA, such as pseudogenes, but there is no firm consensus on the total amount of junk DNA.
A microsatellite is a tract of repetitive DNA in which certain DNA motifs are repeated, typically 5–50 times. Microsatellites occur at thousands of locations within an organism's genome. They have a higher mutation rate than other areas of DNA leading to high genetic diversity. Microsatellites are often referred to as short tandem repeats (STRs) by forensic geneticists and in genetic genealogy, or as simple sequence repeats (SSRs) by plant geneticists.
Gene knockouts are a widely used genetic engineering technique that involves the targeted removal or inactivation of a specific gene within an organism's genome. This can be done through a variety of methods, including homologous recombination, CRISPR-Cas9, and TALENs.

Molecular genetics is a branch of biology that addresses how differences in the structures or expression of DNA molecules manifests as variation among organisms. Molecular genetics often applies an "investigative approach" to determine the structure and/or function of genes in an organism's genome using genetic screens.
A genetic screen or mutagenesis screen is an experimental technique used to identify and select individuals who possess a phenotype of interest in a mutagenized population. Hence a genetic screen is a type of phenotypic screen. Genetic screens can provide important information on gene function as well as the molecular events that underlie a biological process or pathway. While genome projects have identified an extensive inventory of genes in many different organisms, genetic screens can provide valuable insight as to how those genes function.

In genetics and bioinformatics, a single-nucleotide polymorphism is a germline substitution of a single nucleotide at a specific position in the genome that is present in a sufficiently large fraction of considered population.

A frameshift mutation is a genetic mutation caused by indels of a number of nucleotides in a DNA sequence that is not divisible by three. Due to the triplet nature of gene expression by codons, the insertion or deletion can change the reading frame, resulting in a completely different translation from the original. The earlier in the sequence the deletion or insertion occurs, the more altered the protein. A frameshift mutation is not the same as a single-nucleotide polymorphism in which a nucleotide is replaced, rather than inserted or deleted. A frameshift mutation will in general cause the reading of the codons after the mutation to code for different amino acids. The frameshift mutation will also alter the first stop codon encountered in the sequence. The polypeptide being created could be abnormally short or abnormally long, and will most likely not be functional.

Functional genomics is a field of molecular biology that attempts to describe gene functions and interactions. Functional genomics make use of the vast data generated by genomic and transcriptomic projects. Functional genomics focuses on the dynamic aspects such as gene transcription, translation, regulation of gene expression and protein–protein interactions, as opposed to the static aspects of the genomic information such as DNA sequence or structures. A key characteristic of functional genomics studies is their genome-wide approach to these questions, generally involving high-throughput methods rather than a more traditional "candidate-gene" approach.

Loss of heterozygosity (LOH) is a type of genetic abnormality in diploid organisms in which one copy of an entire gene and its surrounding chromosomal region are lost. Since diploid cells have two copies of their genes, one from each parent, a single copy of the lost gene still remains when this happens, but any heterozygosity is no longer present.
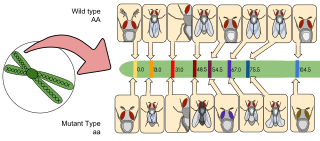
Gene mapping or genome mapping describes the methods used to identify the location of a gene on a chromosome and the distances between genes. Gene mapping can also describe the distances between different sites within a gene.

In population genetics, an ancestry-informative marker (AIM) is a single-nucleotide polymorphism that exhibits substantially different frequencies between different populations. A set of many AIMs can be used to estimate the proportion of ancestry of an individual derived from each population.

Oncogenomics is a sub-field of genomics that characterizes cancer-associated genes. It focuses on genomic, epigenomic and transcript alterations in cancer.

The 1000 Genomes Project, launched in January 2008, was an international research effort to establish by far the most detailed catalogue of human genetic variation. Scientists planned to sequence the genomes of at least one thousand anonymous participants from a number of different ethnic groups within the following three years, using newly developed technologies which were faster and less expensive. In 2010, the project finished its pilot phase, which was described in detail in a publication in the journal Nature. In 2012, the sequencing of 1092 genomes was announced in a Nature publication. In 2015, two papers in Nature reported results and the completion of the project and opportunities for future research.
Methylated DNA immunoprecipitation is a large-scale purification technique in molecular biology that is used to enrich for methylated DNA sequences. It consists of isolating methylated DNA fragments via an antibody raised against 5-methylcytosine (5mC). This technique was first described by Weber M. et al. in 2005 and has helped pave the way for viable methylome-level assessment efforts, as the purified fraction of methylated DNA can be input to high-throughput DNA detection methods such as high-resolution DNA microarrays (MeDIP-chip) or next-generation sequencing (MeDIP-seq). Nonetheless, understanding of the methylome remains rudimentary; its study is complicated by the fact that, like other epigenetic properties, patterns vary from cell-type to cell-type.
Virtual karyotype is the digital information reflecting a karyotype, resulting from the analysis of short sequences of DNA from specific loci all over the genome, which are isolated and enumerated. It detects genomic copy number variations at a higher resolution for level than conventional karyotyping or chromosome-based comparative genomic hybridization (CGH). The main methods used for creating virtual karyotypes are array-comparative genomic hybridization and SNP arrays.
Molecular Inversion Probe (MIP) belongs to the class of Capture by Circularization molecular techniques for performing genomic partitioning, a process through which one captures and enriches specific regions of the genome. Probes used in this technique are single stranded DNA molecules and, similar to other genomic partitioning techniques, contain sequences that are complementary to the target in the genome; these probes hybridize to and capture the genomic target. MIP stands unique from other genomic partitioning strategies in that MIP probes share the common design of two genomic target complementary segments separated by a linker region. With this design, when the probe hybridizes to the target, it undergoes an inversion in configuration and circularizes. Specifically, the two target complementary regions at the 5’ and 3’ ends of the probe become adjacent to one another while the internal linker region forms a free hanging loop. The technology has been used extensively in the HapMap project for large-scale SNP genotyping as well as for studying gene copy alterations and characteristics of specific genomic loci to identify biomarkers for different diseases such as cancer. Key strengths of the MIP technology include its high specificity to the target and its scalability for high-throughput, multiplexed analyses where tens of thousands of genomic loci are assayed simultaneously.

Exome sequencing, also known as whole exome sequencing (WES), is a genomic technique for sequencing all of the protein-coding regions of genes in a genome. It consists of two steps: the first step is to select only the subset of DNA that encodes proteins. These regions are known as exons—humans have about 180,000 exons, constituting about 1% of the human genome, or approximately 30 million base pairs. The second step is to sequence the exonic DNA using any high-throughput DNA sequencing technology.
Bulked segregant analysis (BSA) is a technique used to identify genetic markers associated with a mutant phenotype. This allows geneticists to discover genes conferring certain traits of interest, such as disease resistance or susceptibility.
