Pharmacovigilance, also known as drug safety, is the pharmaceutical science relating to the "collection, detection, assessment, monitoring, and prevention" of adverse effects with pharmaceutical products.
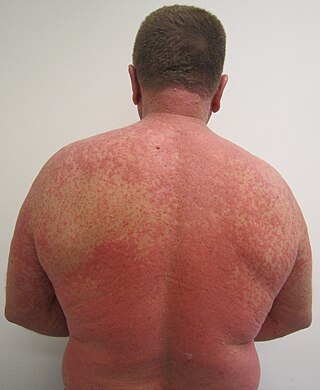
An adverse drug reaction (ADR) is a harmful, unintended result caused by taking medication. ADRs may occur following a single dose or prolonged administration of a drug or may result from the combination of two or more drugs. The meaning of this term differs from the term "side effect" because side effects can be beneficial as well as detrimental. The study of ADRs is the concern of the field known as pharmacovigilance. An adverse event (AE) refers to any unexpected and inappropriate occurrence at the time a drug is used, whether or not the event is associated with the administration of the drug. An ADR is a special type of AE in which a causative relationship can be shown. ADRs are only one type of medication-related harm. Another type of medication-related harm type includes not taking prescribed medications, known as non-adherence. Non-adherence to medications can lead to death and other negative outcomes. Adverse drug reactions require the use of a medication.

The European Medicines Agency (EMA) is an agency of the European Union (EU) in charge of the evaluation and supervision of pharmaceutical products. Prior to 2004, it was known as the European Agency for the Evaluation of Medicinal Products or European Medicines Evaluation Agency (EMEA).
An adverse effect is an undesired harmful effect resulting from a medication or other intervention, such as surgery. An adverse effect may be termed a "side effect", when judged to be secondary to a main or therapeutic effect. The term complication is similar to adverse effect, but the latter is typically used in pharmacological contexts, or when the negative effect is expected or common. If the negative effect results from an unsuitable or incorrect dosage or procedure, this is called a medical error and not an adverse effect. Adverse effects are sometimes referred to as "iatrogenic" because they are generated by a physician/treatment. Some adverse effects occur only when starting, increasing or discontinuing a treatment. Using a drug or other medical intervention which is contraindicated may increase the risk of adverse effects. Adverse effects may cause complications of a disease or procedure and negatively affect its prognosis. They may also lead to non-compliance with a treatment regimen. Adverse effects of medical treatment resulted in 142,000 deaths in 2013 up from 94,000 deaths in 1990 globally.
In pharmaceuticals, an adverse event (AE) is any untoward medical occurrence in a patient or clinical investigation subject administered a pharmaceutical product and which does not necessarily have a causal relationship with this treatment. An adverse event can therefore be any unfavourable and unintended symptom or sign or disease temporally associated with the use of a medicinal (investigational) product, whether or not related to the medicinal (investigational) product.
In drug development, serious adverse event (SAE) is defined as any untoward medical occurrence during a human drug trial that at any dose
- Results in death
- Is life-threatening
- Requires inpatient hospitalization or causes prolongation of existing hospitalization
- Results in persistent or significant disability/incapacity
- May have caused a congenital anomaly/birth defect
- Requires intervention to prevent permanent impairment or damage
In drug development and production, good clinical practice (GCP) is an international quality standard, which governments can then transpose into regulations for clinical trials involving human subjects. GCP follows the International Council for Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH), and enforces tight guidelines on ethical aspects of clinical research.

The Council for International Organizations of Medical Sciences (CIOMS) is an international non-governmental organization of 40 international, national, and associate member groups representing the biomedical science community. It was jointly established by the World Health Organization (WHO) and United Nations Educational, Scientific and Cultural Organization (UNESCO) in 1949 as a successor to the International Medical Congress that organized 17 conferences from 1867 until the 1913 outbreak of World War I.
Veterinary pharmacovigilance in the United Kingdom is overseen by the Veterinary Medicines Directorate (VMD).
A data monitoring committee (DMC) – sometimes called a data and safety monitoring board (DSMB) – is an independent group of experts who monitor patient safety and treatment efficacy data while a clinical trial is ongoing.

Uppsala Monitoring Centre (UMC), located in Uppsala, Sweden, is the field name for the World Health Organization Collaborating Centre for International Drug Monitoring. UMC works by collecting, assessing and communicating information from member countries' national pharmacovigilance centres in regard to the benefits, harm, effectiveness and risks of drugs.
The Yellow Card Scheme is the United Kingdom's system for collecting information on suspected adverse drug reactions (ADRs) to medicines. The scheme allows the safety of the medicines and vaccines that are on the market to be monitored.
Postmarketing surveillance (PMS), also known as post market surveillance, is the practice of monitoring the safety of a pharmaceutical drug or medical device after it has been released on the market and is an important part of the science of pharmacovigilance. Since drugs and medical devices are approved on the basis of clinical trials, which involve relatively small numbers of people who have been selected for this purpose – meaning that they normally do not have other medical conditions which may exist in the general population – postmarketing surveillance can further refine, or confirm or deny, the safety of a drug or device after it is used in the general population by large numbers of people who have a wide variety of medical conditions.

EudraLex is the collection of rules and regulations governing medicinal products in the European Union.
The Clinical Trials Directive is a European Union directive that aimed at facilitating the internal market in medicinal products within the European Union, while at the same time maintaining an appropriate level of protection for public health. It seeks to simplify and harmonise the administrative provisions governing clinical trials in the European Community, by establishing a clear, transparent procedure.
EUDRANET, the European Telecommunication Network in Pharmaceuticals, is an IT platform to facilitate the exchange of information between regulatory partners and industry during submission and evaluation of applications. The aim of EUDRANET is to provide appropriate secure services for inter-Administration data interchange and for exchanges between Administrations and industry. EUDRANET is based on the TESTA backbone infrastructure provided by the IDA Programme.

Research on Adverse Drug Events and Reports (RADAR) is a pharmacovigilance team of 25 doctors who receive calls about possible adverse drug reactions (ADR) and investigate. RADAR is based at Northwestern's Feinberg School of Medicine. RADAR is led by Dennis West. Though it was without funding for its first four years, RADAR has raised about $12 million through grants from the National Institutes of Health, the American Cancer Society and other such institutions. Its work has identified safety problems with 33 drugs. Adverse drug events are a serious health problem.
The following outline is provided as an overview of and topical guide to clinical research:
VigiBase is a World Health Organization's (WHO) global Individual Case Safety Report (ICSR) database that contains ICSRs submitted by the participating member states enrolled under WHO's international drug monitoring programme. It is the single largest drug safety data repository in the world. Since 1978, the Uppsala Monitoring Centre on behalf of WHO, have been maintaining VigiBase.
The Pharmacovigilance Programme of India (PvPI) is an Indian government organization which identifies and responds to drug safety problems. Its activities include receiving reports of adverse drug events and taking necessary action to remedy problems. The Central Drugs Standard Control Organisation established the program in July 2010 with All India Institute of Medical Sciences, New Delhi as the National Coordination Centre, which later shifted to Indian Pharmacopoeia Commission in Ghaziabad on 15 April 2011.