Diagnosis
| | This section is empty. You can help by adding to it. (January 2018) |
| Familial multiple intestinal atresia | |
|---|---|
| Other names | Familial intestinal polyatresia syndrome |
Familial multiple intestinal atresia (FMIA) or familial intestinal polyatresia syndrome (FIPA) is an inherited disorder where atresia occurs at multiple locations throughout the small and large intestines. It presents at birth and the prognosis is very poor with almost all those diagnosed with this condition dying with one month. It may be associated with combined immunodeficiency.
In this disorder lesions can occur anywhere from the stomach to the anus. The clinical presentation depends on the location of the lesions(s).
The underlying lesion in this condition appears to be a mutation in the TTC7A gene. [1] This gene is located on the short arm of chromosome 2 (2p16).
| | This section is empty. You can help by adding to it. (January 2018) |
| | This section is empty. You can help by adding to it. (January 2018) |
This disorder was first described in 1971. [2]

Multiple endocrine neoplasia is a condition which encompasses several distinct syndromes featuring tumors of endocrine glands, each with its own characteristic pattern. In some cases, the tumors are malignant, in others, benign. Benign or malignant tumors of nonendocrine tissues occur as components of some of these tumor syndromes.
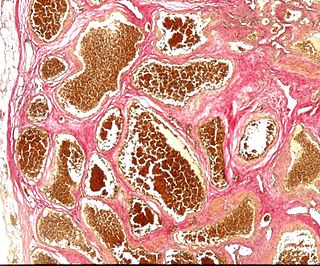
Cerebral Cavernous Malformation (CCM) is a cavernous hemangioma that arises in the central nervous system (CNS). It can be considered to be a variant of hemangioma, and is characterized by grossly large dilated blood vessels and large vascular channels, less well circumscribed, and more involved with deep structures, with a single layer of endothelium and an absence of neuronal tissue within the lesions. These thinly walled vessels resemble sinusoidal cavities filled with stagnant blood. Blood vessels in patients with cerebral cavernous malformations (CCM) can range from a few millimeters to several centimeters in diameter. Most lesions occur in the brain, but any organ may be involved.
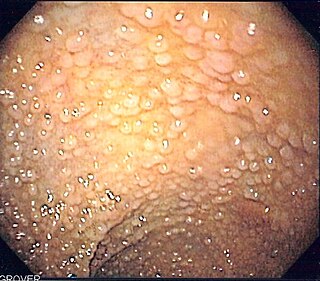
Familial adenomatous polyposis (FAP) is an autosomal dominant inherited condition in which numerous adenomatous polyps form mainly in the epithelium of the large intestine. While these polyps start out benign, malignant transformation into colon cancer occurs when they are left untreated. Three variants are known to exist, FAP and attenuated FAP are caused by APC gene defects on chromosome 5 while autosomal recessive FAP is caused by defects in the MUTYH gene on chromosome 1. Of the three, FAP itself is the most severe and most common; although for all three, the resulting colonic polyps and cancers are initially confined to the colon wall. Detection and removal before metastasis outside the colon can greatly reduce and in many cases eliminate the spread of cancer.

A benign tumor is a mass of cells (tumor) that lacks the ability either to invade neighboring tissue or metastasize. When removed, benign tumors usually do not grow back, whereas malignant tumors are cancerous and sometimes do. Unlike most benign tumors elsewhere in the body, benign brain tumors can be life-threatening. Benign tumors generally have a slower growth rate than malignant tumors and the tumor cells are usually more differentiated. They are typically surrounded by an outer surface or stay contained within the epithelium. Common examples of benign tumors include moles and uterine fibroids.

Alagille syndrome is a genetic disorder that affects primarily the liver and the heart. Problems associated with the disorder generally become evident in infancy or early childhood. The disorder is inherited in an autosomal dominant pattern, and the estimated prevalence of Alagille syndrome is 1 in every 30,000 to 1 in every 40,000 live births. It is named after the French pediatrician Daniel Alagille, who first described the condition in 1969.
Multiple endocrine neoplasia type 1 (MEN-1) is one of a group of disorders, the multiple endocrine neoplasias, that affect the endocrine system through development of neoplastic lesions in pituitary, parathyroid gland and pancreas. It was first described by Paul Wermer in 1954.

Cherubism is a rare genetic disorder that causes prominence in the lower portion in the face. The name is derived from the temporary chubby-cheeked resemblance to putti, often confused with cherubs, in Renaissance paintings.
Vaginal atresia is a condition in which the vagina is abnormally closed or absent. The main causes can either be complete vaginal hypoplasia, or a vaginal obstruction, often caused by an imperforate hymen or, less commonly, a transverse vaginal septum. It results in uterovaginal outflow tract obstruction. This condition does not usually occur by itself within an individual, but coupled with other developmental disorders within the female. The disorders that are usually coupled with a female who has vaginal atresia are Mayer-Rokitansky-Küster-Hauser syndrome, Bardet-Biedl syndrome, or Fraser syndrome. One out of every 5,000 women have this abnormality.

Fundic gland polyposis is a medical syndrome where the fundus and the body of the stomach develop many fundic gland polyps. The condition has been described both in patients with familial adenomatous polyposis (FAP) and attenuated variants (AFAP), and in patients in whom it occurs sporadically.

Intestinal atresia is any congenital malformation of the structure of the intestine that causes bowel obstruction. The malformation can be a narrowing (stenosis), absence or malrotation of a portion of the intestine. These defects can either occur in the small or large intestine.

Antley–Bixler syndrome is a rare, very severe autosomal recessive congenital disorder characterized by malformations and deformities affecting the majority of the skeleton and other areas of the body.

Enteropathy-associated T-cell lymphoma (EATL), previously termed enteropathy-associated T-cell lymphoma, type I and at one time termed enteropathy-type T-cell lymphoma (ETTL), is a complication of coeliac disease in which a malignant T-cell lymphoma develops in areas of the small intestine afflicted by the disease's intense inflammation. While a relatively rare disease, it is the most common type of primary gastrointestinal T-cell lymphoma.
Infantile myofibromatosis (IMF) is a rare tumor found in 1 in 150,000 to 1 in 400,000 live births. It is nonetheless the most common tumor derived from fibrous connective tissue that occurs primarily in infants and young children. IMF tumors are benign in the sense that they do not metastasize to distant tissues although when occurring in the viscera, i.e. internal organs, carry guarded to poor prognoses and can be life-threatening, particularly in newborns and young infants.
Iminoglycinuria is an autosomal recessive disorder of renal tubular transport affecting reabsorption of the amino acid glycine, and the imino acids proline and hydroxyproline. This results in excess urinary excretion of all three acids.

Porokeratosis is a specific disorder of keratinization that is characterized histologically by the presence of a cornoid lamella, a thin column of closely stacked, parakeratotic cells extending through the stratum corneum with a thin or absent granular layer.

Cavernous hemangioma, also called cavernous angioma, venous malformation, cavernoma, or cerebral cavernous malformation (CCM) is a type of venous malformation due to endothelial dysmorphogenesis from a lesion which is present at birth. Despite its designation of a hemangioma, it is not a tumor as it does not display endothelial hyperplasia. The abnormal tissue causes a slowing of blood flow through the cavities, or "caverns". The blood vessels do not form the necessary junctions with surrounding cells, and the structural support from the smooth muscle is hindered, causing leakage into the surrounding tissue. It is the leakage of blood, referred to as hemorrhage, that causes a variety of symptoms known to be associated with the condition.
Congenital tufting enteropathy is an inherited disorder of the small intestine that presents with intractable diarrhea in young children.

Tetratricopeptide repeat domain 7A (TTC7A) is a protein that in humans is encoded by the TTC7A gene.

Strømme syndrome is a very rare autosomal recessive genetic condition characterised by intestinal atresia, eye abnormalities and microcephaly. The intestinal atresia is of the "apple-peel" type, in which the remaining intestine is twisted around its main artery. The front third of the eye is typically underdeveloped, and there is usually moderate developmental delay. Less common features include an atrial septal defect, increased muscle tone or skeletal abnormalities. Physical features may include short stature, large, low-set ears, a small jaw, a large mouth, epicanthic folds, or fine, sparse hair.
CYLD cutaneous syndrome (CCS) is the recently designated term for three rare inherited cutaneous adnexal tumor syndromes: multiple familial trichoepithelioma (MFT1), Brooke–Spiegler syndrome (BSS), and familial cylindromatosis (FC). Cutaneous adnexal tumors are a large group of skin tumors that consist of tissues that have differentiated towards one of the four primary adnexal structures found in normal skin: hair follicles, sebaceous sweat glands, apocrine sweat glands, and eccrine sweat glands. CCS tumors are hair follicle tumors.