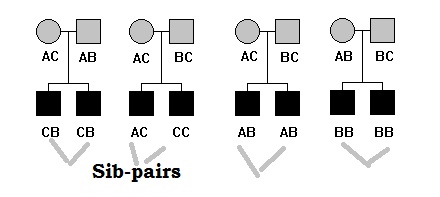
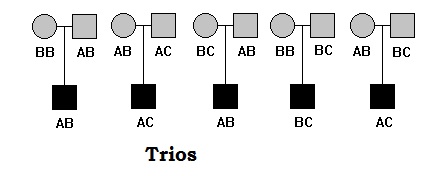
Genetic linkage is the tendency of DNA sequences that are close together on a chromosome to be inherited together during the meiosis phase of sexual reproduction. Two genetic markers that are physically near to each other are unlikely to be separated onto different chromatids during chromosomal crossover, and are therefore said to be more linked than markers that are far apart. In other words, the nearer two genes are on a chromosome, the lower the chance of recombination between them, and the more likely they are to be inherited together. Markers on different chromosomes are perfectly unlinked, although the penetrance of potentially deleterious alleles may be influenced by the presence of other alleles, and these other alleles may be located on other chromosomes than that on which a particular potentially deleterious allele is located.
A quantitative trait locus (QTL) is a locus that correlates with variation of a quantitative trait in the phenotype of a population of organisms. QTLs are mapped by identifying which molecular markers correlate with an observed trait. This is often an early step in identifying the actual genes that cause the trait variation.
In population genetics, linkage disequilibrium (LD) is a measure of non-random association between segments of DNA (alleles) at different positions on the chromosome (loci) in a given population based on a comparison between the frequency at which two alleles are detected together at the same loci versus the frequencies at which each allele is simply detected at that same loci. Loci are said to be in linkage disequilibrium when the frequency of being detected together is higher or lower than expected if the loci were independent and associated randomly.
A polygene is a member of a group of non-epistatic genes that interact additively to influence a phenotypic trait, thus contributing to multiple-gene inheritance, a type of non-Mendelian inheritance, as opposed to single-gene inheritance, which is the core notion of Mendelian inheritance. The term "monozygous" is usually used to refer to a hypothetical gene as it is often difficult to distinguish the effect of an individual gene from the effects of other genes and the environment on a particular phenotype. Advances in statistical methodology and high throughput sequencing are, however, allowing researchers to locate candidate genes for the trait. In the case that such a gene is identified, it is referred to as a quantitative trait locus (QTL). These genes are generally pleiotropic as well. The genes that contribute to type 2 diabetes are thought to be mostly polygenes. In July 2016, scientists reported identifying a set of 355 genes from the last universal common ancestor (LUCA) of all organisms living on Earth.
Genetic association is when one or more genotypes within a population co-occur with a phenotypic trait more often than would be expected by chance occurrence.

In genetics, a locus is a specific, fixed position on a chromosome where a particular gene or genetic marker is located. Each chromosome carries many genes, with each gene occupying a different position or locus; in humans, the total number of protein-coding genes in a complete haploid set of 23 chromosomes is estimated at 19,000–20,000.
In molecular biology and other fields, a molecular marker is a molecule, sampled from some source, that gives information about its source. For example, DNA is a molecular marker that gives information about the organism from which it was taken. For another example, some proteins can be molecular markers of Alzheimer's disease in a person from which they are taken. Molecular markers may be non-biological. Non-biological markers are often used in environmental studies.
A tag SNP is a representative single nucleotide polymorphism (SNP) in a region of the genome with high linkage disequilibrium that represents a group of SNPs called a haplotype. It is possible to identify genetic variation and association to phenotypes without genotyping every SNP in a chromosomal region. This reduces the expense and time of mapping genome areas associated with disease, since it eliminates the need to study every individual SNP. Tag SNPs are useful in whole-genome SNP association studies in which hundreds of thousands of SNPs across the entire genome are genotyped.
In genetics, completelinkage is defined as the state in which two loci are so close together that alleles of these loci are virtually never separated by crossing over. The closer the physical location of two genes on the DNA, the less likely they are to be separated by a crossing-over event. In the case of male Drosophila there is complete absence of recombinant types due to absence of crossing over. This means that all of the genes that start out on a single chromosome, will end up on that same chromosome in their original configuration. In the absence of recombination, only parental phenotypes are expected.
Marker assisted selection or marker aided selection (MAS) is an indirect selection process where a trait of interest is selected based on a marker linked to a trait of interest, rather than on the trait itself. This process has been extensively researched and proposed for plant- and animal- breeding.

In genomics, a genome-wide association study, is an observational study of a genome-wide set of genetic variants in different individuals to see if any variant is associated with a trait. GWA studies typically focus on associations between single-nucleotide polymorphisms (SNPs) and traits like major human diseases, but can equally be applied to any other genetic variants and any other organisms.
A doubled haploid (DH) is a genotype formed when haploid cells undergo chromosome doubling. Artificial production of doubled haploids is important in plant breeding.
An expression quantitative trait locus (eQTL) is a type of quantitative trait locus (QTL), a genomic locus that is associated with phenotypic variation for a specific, quantifiable trait. While the term QTL can refer to a wide range of phenotypic traits, the more specific eQTL refers to traits measured by gene expression, such as mRNA levels. Although named "expression QTLs", not all measures of gene expression can be used for eQTLs. For example, traits quantified by protein levels are instead referred to as protein QTLs (pQTLs).
In genetics, association mapping, also known as "linkage disequilibrium mapping", is a method of mapping quantitative trait loci (QTLs) that takes advantage of historic linkage disequilibrium to link phenotypes to genotypes, uncovering genetic associations.
Nested association mapping (NAM) is a technique designed by the labs of Edward Buckler, James Holland, and Michael McMullen for identifying and dissecting the genetic architecture of complex traits in corn. It is important to note that nested association mapping is a specific technique that cannot be performed outside of a specifically designed population such as the Maize NAM population, the details of which are described below.
In statistical genetics, inclusive composite interval mapping (ICIM) has been proposed as an approach to QTL mapping for populations derived from bi-parental crosses. QTL mapping is based on genetic linkage map and phenotypic data to attempt to locate individual genetic factors on chromosomes and to estimate their genetic effects.
Molecular breeding is the application of molecular biology tools, often in plant breeding and animal breeding. In the broad sense, molecular breeding can be defined as the use of genetic manipulation performed at the level of DNA to improve traits of interest in plants and animals, and it may also include genetic engineering or gene manipulation, molecular marker-assisted selection, and genomic selection. More often, however, molecular breeding implies molecular marker-assisted breeding (MAB) and is defined as the application of molecular biotechnologies, specifically molecular markers, in combination with linkage maps and genomics, to alter and improve plant or animal traits on the basis of genotypic assays.
Mega2 is a data manipulation software for applied statistical genetics. Mega is an acronym for Manipulation Environment for Genetic Analysis.

Complex traits are phenotypes that are controlled by two or more genes and do not follow Mendel's Law of Dominance. They may have a range of expression which is typically continuous. Both environmental and genetic factors often impact the variation in expression. Human height is a continuous trait meaning that there is a wide range of heights. There are an estimated 50 genes that affect the height of a human. Environmental factors, like nutrition, also play a role in a human's height. Other examples of complex traits include: crop yield, plant color, and many diseases including diabetes and Parkinson's disease. One major goal of genetic research today is to better understand the molecular mechanisms through which genetic variants act to influence complex traits. Complex traits are also known as polygenic traits and multigenic traits.
Rohan L. Fernando is a Sri Lankan American geneticist who is a professor of quantitative genetics in the Department of Animal Science at Iowa State University (ISU), US. Fernando's efforts have focused primarily on theory and methods for use of genetic markers in breeding, theory and methods for genetic evaluations of crossbred animals, methodology related to the estimation of genetic parameters and the prediction of genetic merit in populations undergoing selection and non-random mating, Bayesian methodology for analysis of unbalanced mixed model data, optimization of breeding programs, and use of computer simulation to study dynamics of genetic system.