Related Research Articles

Bioinformatics is an interdisciplinary field of science that develops methods and software tools for understanding biological data, especially when the data sets are large and complex. Bioinformatics uses biology, chemistry, physics, computer science, computer programming, information engineering, mathematics and statistics to analyze and interpret biological data. The subsequent process of analyzing and interpreting data is referred to as computational biology.

Genomics is an interdisciplinary field of molecular biology focusing on the structure, function, evolution, mapping, and editing of genomes. A genome is an organism's complete set of DNA, including all of its genes as well as its hierarchical, three-dimensional structural configuration. In contrast to genetics, which refers to the study of individual genes and their roles in inheritance, genomics aims at the collective characterization and quantification of all of an organism's genes, their interrelations and influence on the organism. Genes may direct the production of proteins with the assistance of enzymes and messenger molecules. In turn, proteins make up body structures such as organs and tissues as well as control chemical reactions and carry signals between cells. Genomics also involves the sequencing and analysis of genomes through uses of high throughput DNA sequencing and bioinformatics to assemble and analyze the function and structure of entire genomes. Advances in genomics have triggered a revolution in discovery-based research and systems biology to facilitate understanding of even the most complex biological systems such as the brain.
Metagenomics is the study of genetic material recovered directly from environmental or clinical samples by a method called sequencing. The broad field may also be referred to as environmental genomics, ecogenomics, community genomics or microbiomics.
Metabolic network modelling, also known as metabolic network reconstruction or metabolic pathway analysis, allows for an in-depth insight into the molecular mechanisms of a particular organism. In particular, these models correlate the genome with molecular physiology. A reconstruction breaks down metabolic pathways into their respective reactions and enzymes, and analyzes them within the perspective of the entire network. In simplified terms, a reconstruction collects all of the relevant metabolic information of an organism and compiles it in a mathematical model. Validation and analysis of reconstructions can allow identification of key features of metabolism such as growth yield, resource distribution, network robustness, and gene essentiality. This knowledge can then be applied to create novel biotechnology.

The Joint Genome Institute (JGI) is a scientific user facility for integrative genomic science at Lawrence Berkeley National Laboratory. The mission of the JGI is to advance genomics research in support of the United States Department of Energy's (DOE) missions of energy and the environment. It is one of three national scientific user facilities supported by the Office of Biological and Environmental Research (BER) within the Department of Energy's Office of Research. These BER facilities are part of a more extensive network of 28 national scientific user facilities that operate at the DOE national laboratories.

The Integrated Microbial Genomes system is a genome browsing and annotation platform developed by the U.S. Department of Energy (DOE)-Joint Genome Institute. IMG contains all the draft and complete microbial genomes sequenced by the DOE-JGI integrated with other publicly available genomes. IMG provides users a set of tools for comparative analysis of microbial genomes along three dimensions: genes, genomes and functions. Users can select and transfer them in the comparative analysis carts based upon a variety of criteria. IMG also includes a genome annotation pipeline that integrates information from several tools, including KEGG, Pfam, InterPro, and the Gene Ontology, among others. Users can also type or upload their own gene annotations and the IMG system will allow them to generate Genbank or EMBL format files containing these annotations.

MicrobesOnline is a publicly and freely accessible website that hosts multiple comparative genomic tools for comparing microbial species at the genomic, transcriptomic and functional levels. MicrobesOnline was developed by the Virtual Institute for Microbial Stress and Survival, which is based at the Lawrence Berkeley National Laboratory in Berkeley, California. The site was launched in 2005, with regular updates until 2011.
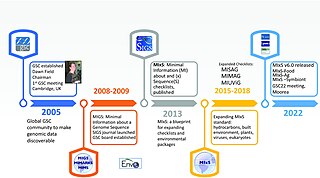
The Genomic Standards Consortium (GSC) is an initiative working towards richer descriptions of our collection of genomes, metagenomes and marker genes. Established in September 2005, this international community includes representatives from a range of major sequencing and bioinformatics centres and research institutions. The goal of the GSC is to promote mechanisms for standardizing the description of (meta)genomes, including the exchange and integration of (meta)genomic data. The number and pace of genomic and metagenomic sequencing projects will only increase as the use of ultra-high-throughput methods becomes common place and standards are vital to scientific progress and data sharing.
The Human Microbiome Project (HMP) was a United States National Institutes of Health (NIH) research initiative to improve understanding of the microbiota involved in human health and disease. Launched in 2007, the first phase (HMP1) focused on identifying and characterizing human microbiota. The second phase, known as the Integrative Human Microbiome Project (iHMP) launched in 2014 with the aim of generating resources to characterize the microbiome and elucidating the roles of microbes in health and disease states. The program received $170 million in funding by the NIH Common Fund from 2007 to 2016.
The Variant Call Format or VCF is a standard text file format used in bioinformatics for storing gene sequence variations. The format was developed in 2010 for the 1000 Genomes Project and has since been used by other large-scale genotyping and DNA sequencing projects. VCF is a common output format for variant calling programs due to its relative simplicity and scalability. Many tools have been developed for editing and manipulating VCF files, including VCFtools, which was released in conjunction with the VCF format in 2011, and BCFtools, which was included as part of SAMtools until being split into an independent package in 2014.

The Sequence Read Archive is a bioinformatics database that provides a public repository for DNA sequencing data, especially the "short reads" generated by high-throughput sequencing, which are typically less than 1,000 base pairs in length. The archive is part of the International Nucleotide Sequence Database Collaboration (INSDC), and run as a collaboration between the NCBI, the European Bioinformatics Institute (EBI), and the DNA Data Bank of Japan (DDBJ).
High-throughput sequencing technologies have led to a dramatic decline of genome sequencing costs and to an astonishingly rapid accumulation of genomic data. These technologies are enabling ambitious genome sequencing endeavours, such as the 1000 Genomes Project and 1001 Genomes Project. The storage and transfer of the tremendous amount of genomic data have become a mainstream problem, motivating the development of high-performance compression tools designed specifically for genomic data. A recent surge of interest in the development of novel algorithms and tools for storing and managing genomic re-sequencing data emphasizes the growing demand for efficient methods for genomic data compression.

The Earth Microbiome Project (EMP) is an initiative founded by Janet Jansson, Jack Gilbert and Rob Knight in 2010 to collect natural samples and to analyze the microbial community around the globe.
The genomic epidemiological database for global identification of microorganisms or global microbial identifier is a platform for storing whole genome sequencing data of microorganisms, for the identification of relevant genes and for the comparison of genomes to detect and track-and-trace infectious disease outbreaks and emerging pathogens. The database holds two types of information: 1) genomic information of microorganisms, linked to, 2) metadata of those microorganism such as epidemiological details. The database includes all genera of microorganisms: bacteria, viruses, parasites and fungi.
MG-RAST, an open-source web application server, facilitates automatic phylogenetic and functional analysis of metagenomes. It stands as one of the largest repositories for metagenomic data, employing the acronym for Metagenomic Rapid Annotations using Subsystems Technology (MG-RAST). This platform utilizes a pipeline that automatically assigns functions to metagenomic sequences, conducting sequence comparisons at both nucleotide and amino acid levels. Users benefit from phylogenetic and functional insights into the analyzed metagenomes, along with tools for comparing different datasets. MG-RAST also offers a RESTful API for programmatic access.

Viral metagenomics uses metagenomic technologies to detect viral genomic material from diverse environmental and clinical samples. Viruses are the most abundant biological entity and are extremely diverse; however, only a small fraction of viruses have been sequenced and only an even smaller fraction have been isolated and cultured. Sequencing viruses can be challenging because viruses lack a universally conserved marker gene so gene-based approaches are limited. Metagenomics can be used to study and analyze unculturable viruses and has been an important tool in understanding viral diversity and abundance and in the discovery of novel viruses. For example, metagenomics methods have been used to describe viruses associated with cancerous tumors and in terrestrial ecosystems.
Deinococcus frigens is a species of low temperature and drought-tolerating, UV-resistant bacteria from Antarctica. It is Gram-positive, non-motile and coccoid-shaped. Its type strain is AA-692. Individual Deinococcus frigens range in size from 0.9-2.0 μm and colonies appear orange or pink in color. Liquid-grown cells viewed using phase-contrast light microscopy and transmission electron microscopy on agar-coated slides show that isolated D. frigens appear to produce buds. Comparison of the genomes of Deiococcus radiodurans and D. frigens have predicted that no flagellar assembly exists in D. frigens.
Nikos Kyrpides is a Greek-American bioscientist who has worked on the origins of life, information processing, bioinformatics, microbiology, metagenomics and microbiome data science. He is a senior staff scientist at the Berkeley National Laboratory, head of the Prokaryote Super Program and leads the Microbiome Data Science program at the US Department of Energy Joint Genome Institute.
Genome mining describes the exploitation of genomic information for the discovery of biosynthetic pathways of natural products and their possible interactions. It depends on computational technology and bioinformatics tools. The mining process relies on a huge amount of data accessible in genomic databases. By applying data mining algorithms, the data can be used to generate new knowledge in several areas of medicinal chemistry, such as discovering novel natural products.
References
- 1 2 Kyrpides NC (September 1999). "Genomes OnLine Database (GOLD 1.0): a monitor of complete and ongoing genome projects world-wide". Bioinformatics. 15 (9): 773–4. doi: 10.1093/bioinformatics/15.9.773 . PMID 10498782.
- 1 2 3 Reddy, T. B. K.; Thomas, A. D.; Stamatis, D.; Bertsch, J.; Isbandi, M.; Jansson, J.; Mallajosyula, J.; Pagani, I.; Lobos, E. A.; Kyrpides, N. C. (27 October 2014). "The Genomes OnLine Database (GOLD) v.5: a metadata management system based on a four level (meta)genome project classification". Nucleic Acids Research. 43 (D1): D1099–D1106. doi:10.1093/nar/gku950. PMC 4384021 . PMID 25348402.
- ↑ "JGI GOLD - Home". Archived from the original on 16 April 2021. Retrieved 5 August 2015.